Command Palette
Search for a command to run...
機械学習による予測誤差は、DFTの精度を上回る
機械学習による予測誤差は、DFTの精度を上回る
Felix A. Faber Luke Hutchison Bing Huang Justin Gilmer Samuel S. Schoenholz George E. Dahl Oriol Vinyals Steven Kearnes Patrick F. Riley O. Anatole von Lilienfeld
概要
有機分子の電子基底状態に関する13の物性に対する高速な機械学習(ML)モデル構築において、回帰器および分子表現の選択がもたらす影響を検討した。各回帰器/表現/物性の組み合わせの性能は、最大約117,000種類の異なる分子を用いた学習曲線を通じて評価された。訓練およびテストに用いた分子構造および物性データは、ハイブリッド密度汎関数理論(DFT)レベルでの計算結果であり、QM9データベース[Ramakrishnanら、{em Scientific Data} {bf 1}, 140022(2014)]から取得したものである。対象となる物性には、電気双極子モーメント、極化率、HOMO/LUMOエネルギーおよびバンドギャップ、電子空間拡がり、ゼロ点振動エネルギー、原子化エンタルピーおよび自由エネルギー、熱容量、および最高の基底振動周波数が含まれる。文献に報告されたさまざまな分子表現(コロンブ行列、ボックス・オブ・ボンズ、BAMLおよびECFP4、分子グラフ(MG))に加え、新たに開発された分布に基づく表現(距離のヒストグラム(HD)、角度のヒストグラム(HDA/MARAD)、二面角のヒストグラム(HDAD))も検討した。回帰器としては、線形モデル(ベイズリッジ回帰(BR)、エラスティックネット正則化付き線形回帰(EN))、ランダムフォレスト(RF)、カーネルリッジ回帰(KRR)、および2種類のニューラルネットワーク(グラフ畳み込み(GC)、ゲート付きグラフネットワーク(GG))を用いた。数値的な証拠として、すべての物性について、MLモデルの予測値がDFT値と比べて、DFT値と実験値のずれよりも小さくなることが示された。さらに、ハイブリッドDFTを基準として用いた際の、検証データに対する予測誤差は、化学的精度(chemical accuracy)と同等、あるいはそれと近い水準に達している。これらの結果から、明示的に電子相関を考慮した量子計算データまたは実験データが利用可能であれば、MLモデルはハイブリッドDFTよりも高い精度を達成できる可能性が示唆される。
One-sentence Summary
The authors, from the University of Basel and Google (Mountain View and London), demonstrate that machine learning models—specifically molecular graph-based neural networks (MG/GC, MG/GG) and histogram-based representations (HDAD) with kernel ridge regression—achieve out-of-sample prediction errors on par with or below hybrid DFT accuracy for 13 electronic and energetic properties of organic molecules, with several reaching chemical accuracy, suggesting ML could surpass DFT if trained on higher-level reference data.
Key Contributions
-
The study systematically evaluates machine learning models for predicting 13 electronic and energetic properties of organic molecules using the QM9 dataset, with training sets approaching 117,000 molecules, to assess whether ML can surpass the accuracy of hybrid density functional theory (DFT) in predicting quantum chemical properties.
-
It demonstrates that specific combinations of molecular representations—such as molecular graphs (MG) for electronic properties and histogram-based descriptors (HDAD) for energetic properties—paired with appropriate regressors like graph convolutions (GC) or kernel ridge regression (KRR), achieve out-of-sample errors comparable to or below the estimated error of DFT relative to experiment.
-
For all properties, the best-performing ML models exhibit prediction errors that are either on par with or lower than the typical DFT error relative to experiment, with many reaching chemical accuracy, suggesting that ML models trained on high-level quantum data could outperform DFT if such data were available.
Introduction
The authors investigate machine learning (ML) models for predicting electronic and energetic properties of organic molecules, using the QM9 dataset of over 117,000 molecules with high-accuracy hybrid DFT reference data. This work is significant because it systematically evaluates a wide range of molecular representations and regressors—spanning traditional descriptors like Coulomb matrices and ECFP4 to novel distribution-based features (e.g., HDAD, MARAD) and graph-based models (GC, GG)—to determine optimal combinations for different properties. Prior studies often lacked consistency in data size, representation, or benchmarking scope, making it unclear whether ML could surpass DFT in accuracy. The key contribution is demonstrating that, for all 13 properties studied, the best-performing ML models achieve out-of-sample errors that are smaller than or comparable to the estimated error of hybrid DFT relative to experiment, with many reaching chemical accuracy. This suggests that, given access to higher-level quantum or experimental data, ML models could outperform DFT in predictive accuracy while being orders of magnitude faster.
Dataset
-
The dataset consists of approximately 131,000 drug-like organic molecules from the QM9 dataset, limited to atoms H, C, O, N, and F with up to 9 heavy atoms. It excludes 3,053 molecules failing SMILES consistency checks and two linear molecules.
-
Each molecule includes 13 quantum mechanical properties calculated at the B3LYP/6-31G(2df,p) DFT level: dipole moment, static polarizability, HOMO and LUMO eigenvalues, HOMO-LUMO gap, electronic spatial extent, zero-point vibrational energy, and atomization energies at 0 K (U₀), room temperature (U, H, G), heat capacity, and highest fundamental vibrational frequency. The analysis focuses on U₀ for energy-related properties.
-
The authors use a standard train/validation/test split, with all models trained on the same data partitioning. They evaluate multiple representations—Coulomb Matrix (CM), Bag of Bonds (BoB), BAML (Bonds, Angles, Machine Learning), ECFP4 fingerprints, and molecular graphs (MG)—each processed differently.
-
For CM and BoB, atom ordering is based on the L¹ norm of atom rows, and BoB groups Coulomb terms by atom pairs and sorts them by magnitude. BAML extends BoB by replacing Coulomb terms with Morse/Lennard-Jones potentials for bonded/non-bonded pairs and adding angular and torsional terms based on UFF. ECFP4 uses a fixed-length 1024-bit vector derived from hashed subgraphs up to 4 bonds in diameter, based solely on the molecular graph.
-
Molecular graph (MG) features include atom-level descriptors (e.g., atomic number, hybridization, number of hydrogens) and pair-level features (e.g., bond type, Euclidean distance). Distance information is discretized into 10 bins (0–2, 2–2.5, ..., 6+ Å), and the adjacency matrix uses 14 discrete types (bond types and distance bins). 367 molecules were excluded due to SMILES conversion failures in OpenBabel or RDKit.
-
All property values are standardized to zero mean and unit variance before training. Gated Graph Neural Networks (GG) are trained individually per property, with distance bins incorporated into the adjacency matrix and hidden states updated over multiple steps. Random Forest models use 120 trees and are trained on standardized data.
Method
The authors leverage a suite of molecular representation methods designed to capture structural and geometric information from molecules, each tailored to different modeling paradigms. The framework begins with the Molecular Atomic Radial Angular Distribution (MARAD), a radial distribution function (RDF)-based representation that extends the atomic representation ARAD by incorporating three RDFs: pairwise distances, and parallel and orthogonal projections of distances in atom triplets. While MARAD is inherently distance-based, most regressors—such as Bayesian Ridge Regression (BR), Elastic Net (EN), and Random Forest (RF)—do not rely on inner products or distances between representations. To address this limitation, MARAD is projected onto discrete bins, enabling compatibility with these models. This binning process allows the representation to be used across a broad range of regression techniques, except for Graph Convolution (GC) and Graph Generation (GG), which exclusively use the Molecular Graph (MG) representation.
As an alternative to MARAD, the authors investigate histogram-based representations that directly encode pairwise distances, triple-wise angles, and quad-wise dihedral angles through manually generated bins. These representations, termed HD (Histogram of distances), HDA (Histogram of distances and angles), and HDAD (Histogram of distances, angles, and dihedral angles), are constructed by iterating over each atom ai in a molecule and computing features relative to its neighbors. For distance features, the distance between ai and aj (where i=j) is measured and labeled with the sorted atomic symbols of the pair (with hydrogen listed last). Angle features are derived from the principal angles formed by vectors from ai to its two nearest neighbors, aj and ak, and are labeled with the atomic type of ai followed by the alphabetically sorted types of aj and ak. Dihedral angle features are computed from the principal angles between two planes defined by ai, aj, ak, and al, with the label formed by the atomic symbols of ai, aj, ak, and al, again sorted alphabetically with hydrogens last.
The resulting histograms for each label type are analyzed to identify significant local minima and maxima, which are interpreted as structural commonalities. Bin centers are placed at these extrema, and values at 15–25 such centers are selected as the representation for each label type. For each molecule, the features are rendered into a fixed-size vector through a two-step process: first, binning and interpolation, where each feature value is projected onto the two nearest bins using linear interpolation; second, reduction, where the contributions within each bin are summed to produce a single value. This process ensures that the representation is both compact and informative, capturing key structural motifs.
The models employed include Kernel Ridge Regression (KRR), Bayesian Ridge Regression (BR), and Elastic Net (EN), each with distinct regularization strategies. KRR uses kernel functions as a basis set and predicts a property p of a query molecule m as a weighted sum of kernel functions between m and all training molecules mitrain:
p(m)=i∑NαiK(m,mitrain)where αi are regression coefficients obtained by minimizing the Euclidean distance between predicted and reference properties. The authors use Laplacian and Gaussian kernels, with regularization set to 10−9 due to the low noise in the data. Kernel widths are optimized via screening on a base-2 logarithmic grid, and feature vectors are normalized using the Euclidean norm for the Gaussian kernel and the Manhattan norm for the Laplacian kernel.
BR is a linear model with an L2 penalty on coefficients, where the optimal regularization strength is estimated from the data, eliminating the need for manual hyperparameter tuning. EN combines L1 and L2 penalties, with the relative strength controlled by the l1_ratio hyperparameter. The authors set l1_ratio = 0.5 and perform a hyperparameter search over a base-10 logarithmic grid for the regularization parameter.
Finally, the Graph Convolution (GC) model, based on the architecture described by Kearnes et al., is adapted with three key modifications. First, the "Pair order invariance" property is removed by simplifying the (A→P) transformation, as the model only uses the atom layer for molecule-level features. Second, Euclidean distances between atoms are incorporated into the (P→A) transformation by concatenating the convolution output with scaled versions of itself using d−1, d−2, d−3, and d−6, where d is the distance. Third, molecule-level features are generated by summing softmax-transformed atom layer vectors across atoms, a method inspired by fingerprinting techniques such as Extended Connectivity Fingerprints. This approach is found to perform as well as or better than the Gaussian histograms used in the original GC model. Hyperparameter optimization is conducted using Gaussian Process Bandit Optimization via HyperTune, with the search based on validation set performance for a single data fold.
Experiment
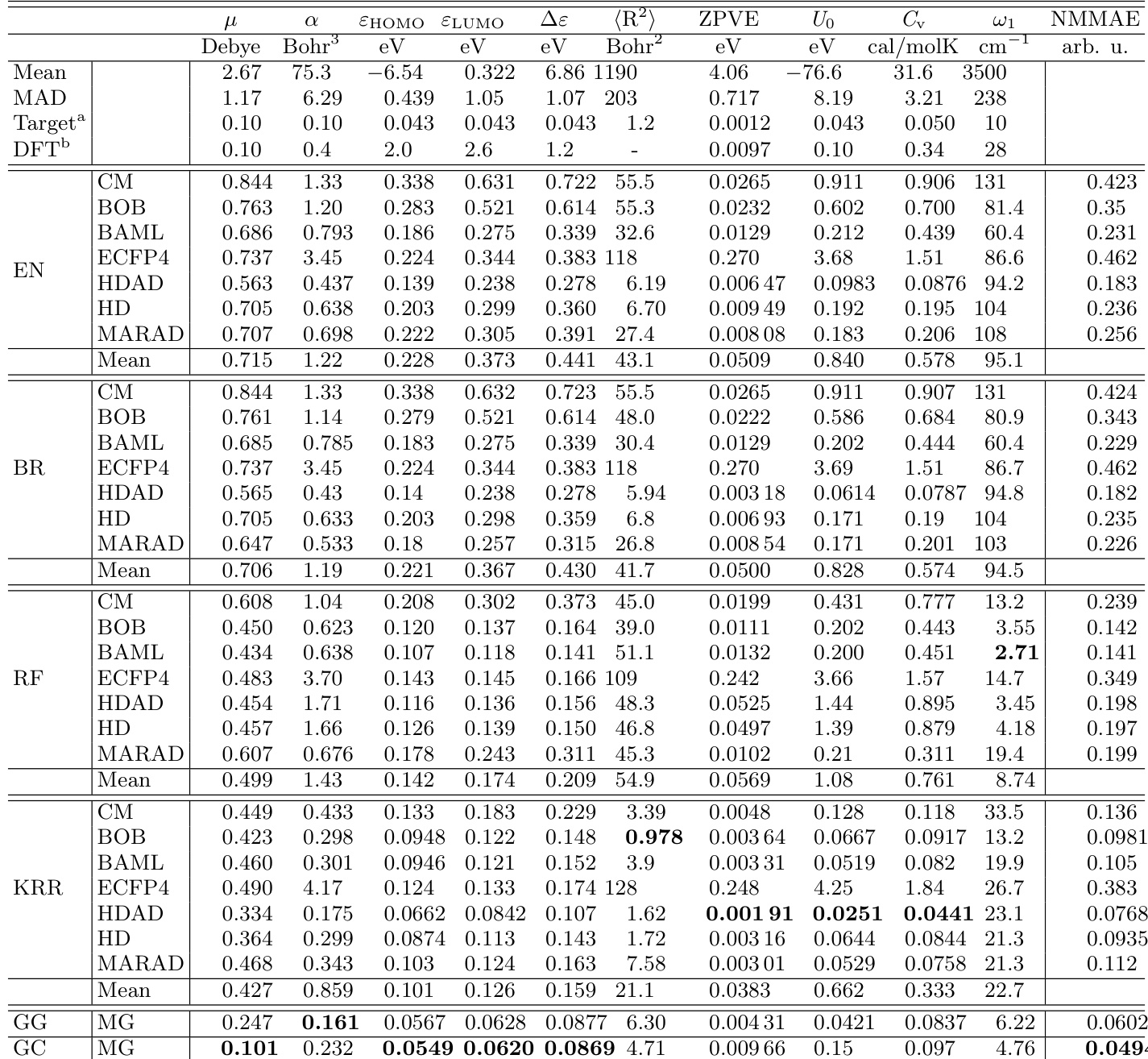
- Evaluated multiple regressor-representation combinations on ~117k molecules from QM9 using 10-fold cross-validation and test set analysis; best models achieved chemical accuracy or better for 8 out of 13 properties, with remaining properties within a factor of 2 of chemical accuracy.
- GC and GG neural networks outperformed other regressors across most properties, especially for electronic properties (μ, α, ε_HOMO, ε_LUMO, Δε); KRR performed strongly for extensive properties (⟨R²⟩, ZPVE, U₀, Cᵥ) and was second-best overall.
- HDAD representation consistently outperformed HD and HDA, particularly with KRR, due to effective capture of three- and four-body interactions; its simplicity and interpretability align with Occam’s razor, though it lacks differentiability.
- RF regressor achieved exceptional performance only for ω₁ (highest vibrational frequency), with errors as low as single-digit cm⁻¹, attributed to its ability to detect bond types via decision trees; however, it underperformed on other properties.
- ECFP4 and BR/EN regressors showed poor generalization; ECFP4 failed on extensive properties, while BR and EN exhibited high errors and limited improvement with training size due to low model flexibility.
- Learning curves demonstrated systematic error reduction with increasing training set size, with steeper slopes for certain property-representation pairs indicating lower effective dimensionality in chemical space.
The authors use learning curves to evaluate the performance of various regressor and representation combinations across multiple molecular properties, showing that model errors generally decrease with increasing training set size. Results show that graph-based neural networks (GC and GG) and kernel ridge regression (KRR) achieve the lowest mean absolute errors for most properties, with GC and GG performing best for electronic properties and HDAD/KRR excelling for extensive properties like polarizability and heat capacity.
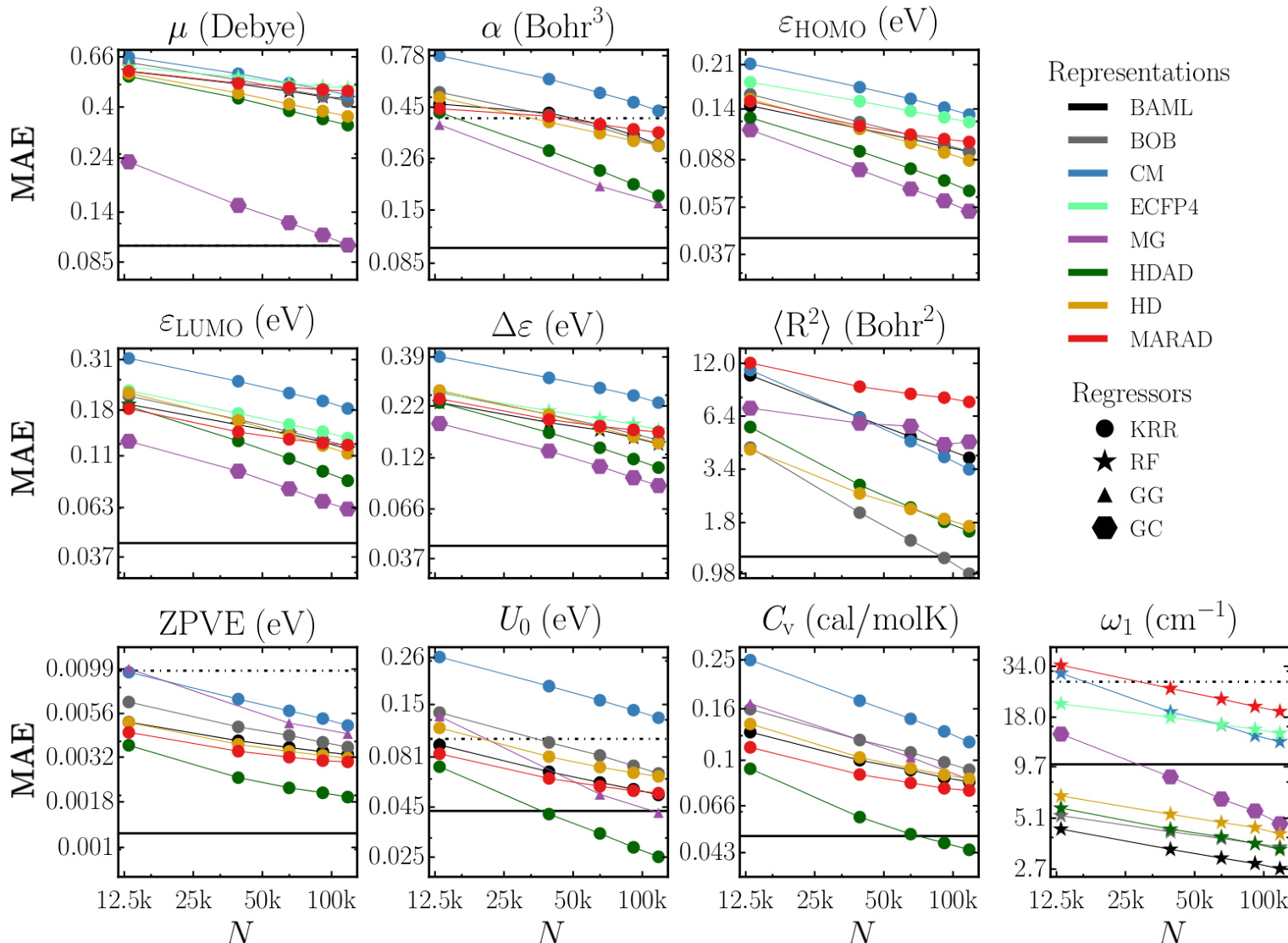
The authors use a comprehensive benchmark of machine learning models on the QM9 dataset to evaluate the performance of various regressors and representations for predicting molecular properties. Results show that graph-based neural networks (GC, GG) and kernel ridge regression (KRR) achieve the lowest errors, with GC and GG outperforming other methods for electronic properties and KRR excelling for extensive properties, while EN, BR, and RF regressors consistently yield higher errors.