Command Palette
Search for a command to run...
Prédictions précises de nouvelles interactions biomoléculaires avec IsoDDE
Prédictions précises de nouvelles interactions biomoléculaires avec IsoDDE
Isomorphic Labs Team
Résumé
La prédiction des interactions biomoléculaires est fondamentale pour la conception rationnelle de médicaments ; cependant, l'atteinte d'une précision expérimentale et d'une capacité de généralisation à travers de nouveaux espaces chimiques demeure un goulot d'étranglement critique. Bien que les approches de deep learning, telles qu'AlphaFold 3, aient fait progresser la prédiction de structures, les benchmarks révèlent que des limites persistent dans la généralisation à des régions inexplorées de l'espace moléculaire, dans l'estimation de l'affinité de liaison et dans la détection de sites de liaison moléculaire sur des surfaces protéiques jusqu'alors non caractérisées.Dans cette étude, nous présentons l'Isomorphic Labs Drug Design Engine (IsoDDE), un système computationnel unifié conçu pour lever ces limitations. Nous démontrons que l'IsoDDE fait plus que doubler la précision d'AlphaFold 3 sur un benchmark de généralisation protéine-ligand complexe, en modélisant avec succès des événements hors distribution (out-of-distribution) tels que l'ajustement induit (induced fits), et en identifiant avec précision de nouvelles poches de liaison.Dans le domaine des produits biologiques, l'IsoDDE surpasse substantiellement les modèles existants, établissant un nouvel état de l'art pour la prédiction de l'interface anticorps-antigène et la modélisation de la boucle CDR-H3. Enfin, pour les lieurs de petites molécules (small molecule binders), les prédictions d'affinité de l'IsoDDE dépassent les méthodes physiques de référence (gold-standard physics-based methods), comblant ainsi l'écart vers une précision de niveau expérimental sans la charge computationnelle des workflows physiques traditionnels. Nos résultats démontrent que l'IsoDDE offre un socle évolutif pour la conception de médicaments assistée par IA, fournissant la fidélité prédictive nécessaire pour naviguer dans de nouveaux systèmes biologiques avec une précision sans précédent.
One-sentence Summary
The Isomorphic Labs Drug Design Engine (IsoDDE) is a unified computational system that outperforms AlphaFold 3 on protein-ligand generalization benchmarks by modeling complex out-of-distribution events like induced fits, achieving state-of-the-art accuracy in antibody-antigen interface and CDR-H3 loop modeling, and providing small molecule binding affinity predictions that exceed gold-standard physics-based methods.
Key Contributions
- The paper introduces the Isomorphic Labs Drug Design Engine (IsoDDE), a unified computational system that achieves a step change in unconditional accuracy for biomolecular interaction prediction. This architecture demonstrates the ability to more than double the accuracy of AlphaFold 3 on protein-ligand generalization benchmarks and successfully models complex out-of-distribution events like induced fits.
- This work presents a high-performance model for the biologics domain that establishes a new state of the art in antibody-antigen interface prediction and CDR-H3 loop modeling. The system also enables blind pocket identification, allowing for the discovery of novel binding sites on previously uncharacterized protein surfaces without requiring a specified ligand.
- The research provides a quantitative binding affinity estimation capability that outperforms gold-standard physics-based methods for small molecule binders. This approach bridges the gap toward experimental-grade precision while avoiding the high computational overhead associated with traditional free energy perturbation workflows.
Introduction
Accurate prediction of biomolecular interactions is essential for rational drug design and the discovery of new biological mechanisms. While deep learning models like AlphaFold 3 have advanced structure prediction, they often struggle to generalize to unexplored chemical spaces, fail to identify novel binding pockets, and lack the ability to provide precise quantitative binding affinity. Current methods for estimating affinity are either computationally expensive physics-based simulations or deep learning models that correlate poorly with experimental data. The authors introduce the Isomorphic Labs Drug Design Engine (IsoDDE), a unified computational system that achieves a step change in accuracy and generalization. IsoDDE more than doubles the accuracy of previous state-of-the-art models on protein-ligand benchmarks, provides superior antibody-antigen interface modeling, and delivers binding affinity predictions that exceed gold-standard physics-based methods.
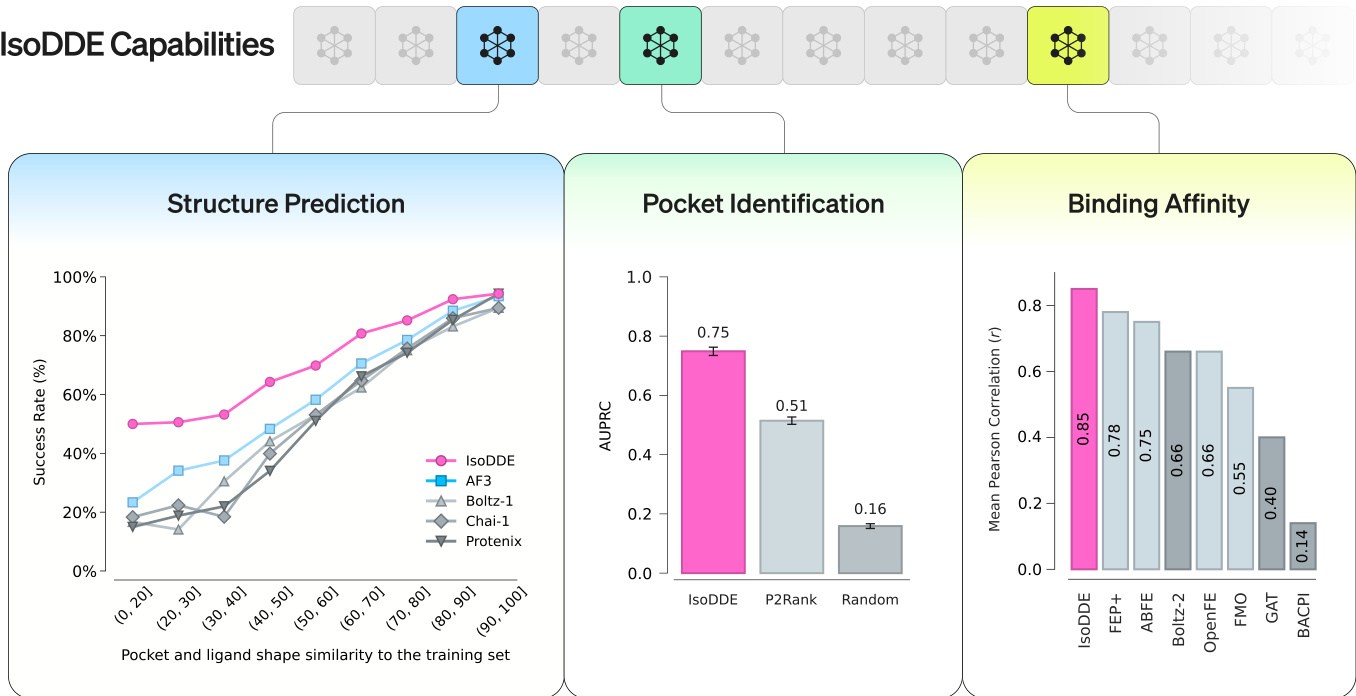
Experiment
IsoDDE was evaluated across multiple benchmarks, including Runs N' Poses, FoldBench, and antibody-antigen datasets, to assess its structural modeling and generalization capabilities. The results demonstrate that the model significantly outperforms previous state-of-the-art methods like AlphaFold 3 and Boltz-2, particularly when predicting highly novel protein-ligand interfaces and complex antibody-antigen structures. Furthermore, IsoDDE shows exceptional performance in predicting binding affinities and identifying cryptic pockets, proving to be a robust tool for navigating diverse chemical and biological spaces in drug discovery.
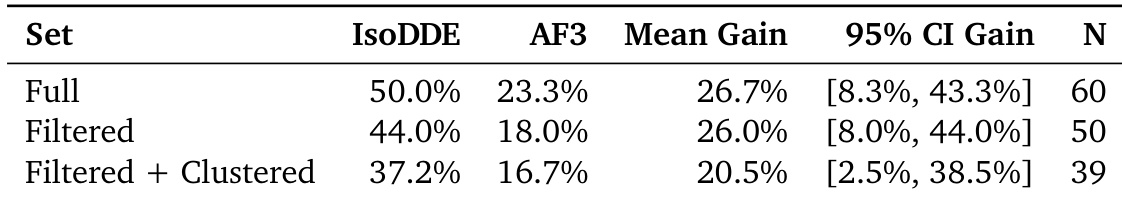
The authors compare the performance of IsoDDE against AF3 across different subsets of the Runs N' Poses benchmark. Results show that IsoDDE achieves a higher success rate than AF3 in the full dataset, the filtered dataset, and the filtered plus clustered dataset. IsoDDE demonstrates a consistent performance advantage over AF3 across all evaluated data subsets The mean gain in success rate remains substantial whether using the full or filtered datasets IsoDDE maintains superior performance even when the data is filtered and clustered to reduce redundancy
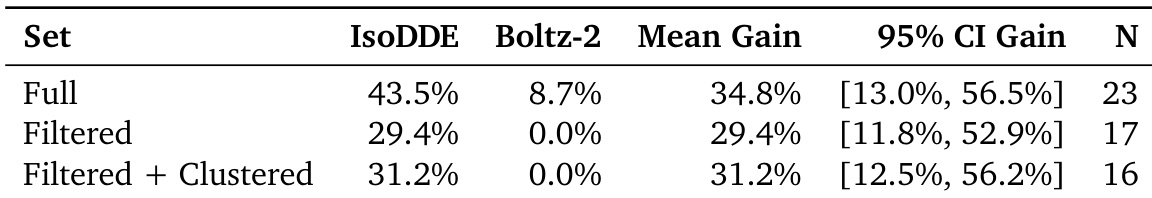
The authors evaluate the performance of IsoDDE against Boltz-2 across different data subsets. Results show that IsoDDE maintains a higher success rate than Boltz-2 in the full, filtered, and clustered datasets. IsoDDE demonstrates a substantial mean gain in performance compared to Boltz-2 across all tested sets The model maintains consistent improvements even when data is filtered or clustered Performance advantages are observed in both the full dataset and more specialized subsets
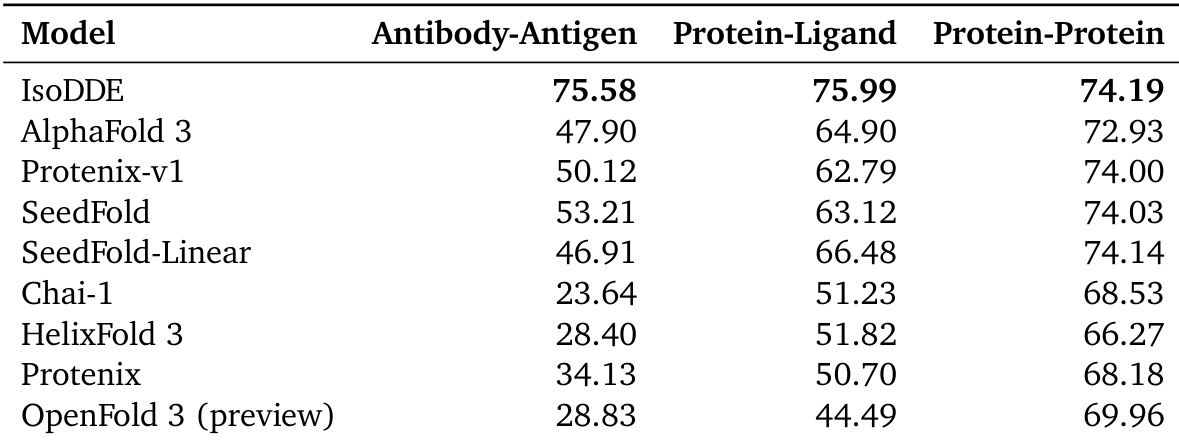
The authors evaluate IsoDDE's structural modeling performance on the FoldBench dataset across three distinct interface types. Results show that IsoDDE achieves superior success rates compared to several external models in antibody-antigen, protein-ligand, and protein-protein benchmarks. IsoDDE demonstrates higher success rates in antibody-antigen interface prediction than all compared models The model shows improved performance in protein-ligand modeling relative to the other evaluated methods IsoDDE maintains a competitive advantage across protein-protein interaction benchmarks
IsoDDE was evaluated against AF3, Boltz-2, and various external models using the Runs N' Poses and FoldBench benchmarks to assess its structural modeling capabilities. Across diverse datasets and interface types, including antibody-antigen, protein-ligand, and protein-protein interactions, IsoDDE consistently demonstrated superior success rates. The results indicate that the model maintains a robust performance advantage even when data is filtered or clustered to reduce redundancy.