Command Palette
Search for a command to run...
Selected for ICLR 2025! Cambridge University Proposed the Celcomen Model, Which Is the First to Achieve Identifiability of Causal Inference in Spatial Transcriptomics Analysis
In biology, the gene expression profile of a cell encodes information about both its intrinsic properties and the external tissue microenvironment. Unraveling the causal relationship between these two effects is critical to fully understand the complex interactions within and between cells. To this end, a robust causal disentanglement framework is needed.
Causal decoupling is a machine learning method that aims to separate useful features from irrelevant features by revealing causal relationships in data, thereby reducing the model's reliance on false associations and improving the model's robustness and generalization capabilities. While machine learning theories such as causal decoupling are developing, technological advances in the field of biology have also promoted the development of spatial transcriptomics, allowing researchers to simultaneously measure cell gene expression and spatial coordinates at single-cell resolution, and to conduct perturbation experiments such as gene knockout on a large scale in spatial samples.
However,Current computational approaches to spatial transcriptomics often neglect modeling of causal perturbations at the cellular and tissue levels.This is crucial for revealing the mechanisms behind tissue disease states. For example, the Virtual Cells model can predict the effects of changes in the microenvironment and macroenvironment (such as donor age, cell tissue, drug treatment, gRNA-mediated gene knockout, etc.) on gene expression, and the Virtual Tissues model can not only estimate the effects of the environment on individual cells, but also infer the effects of individual cells on their surroundings and the overall tissue.
Based on this,A research team from the University of Cambridge proposed a virtual tissue model called Celcomen, which is essentially a new graph neural network based on mathematical causality to unlock the secrets of intracellular and intercellular gene regulation in spatial transcriptomics and single-cell data.The researchers validated Celcomen's ability to unravel and recover gene-gene interactions in both real and self-simulated spatial transcriptomics data.
The related results were selected for ICLR 2025 under the title "Estimation of single-cell and tissue perturbation effect in spatial transcriptomics via Spatial Causal Disentanglement".
Research highlights:
* The study proves the feasibility of extending the virtual cell model to the virtual tissue model
* The study proposes the first causally identifiable model in spatial transcriptomics analysis
* Infer gene regulation by integrating dissociated single-cell data and spatial single-cell data
Paper address:
https://openreview.net/forum?id=Tqdsruwyac
The open source project "awesome-ai4s" brings together more than 200 AI4S paper interpretations and provides massive data sets and tools:
https://github.com/hyperai/awesome-ai4s
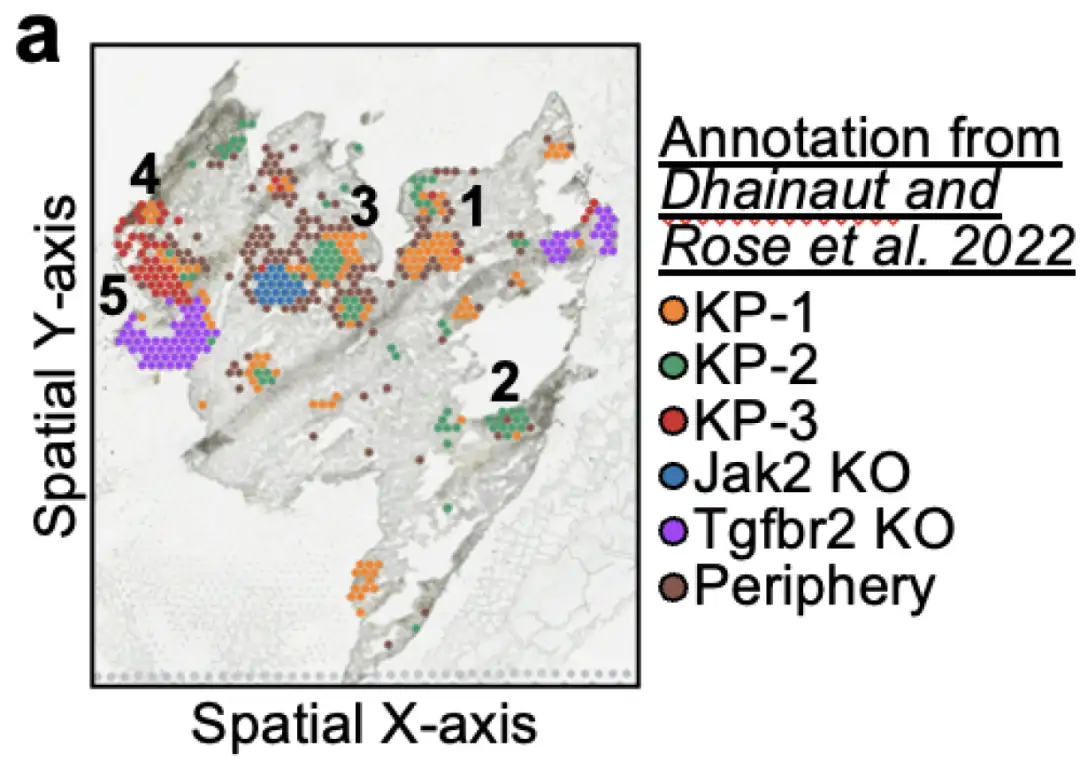
Dataset: First attempt to use the Perturbmap dataset
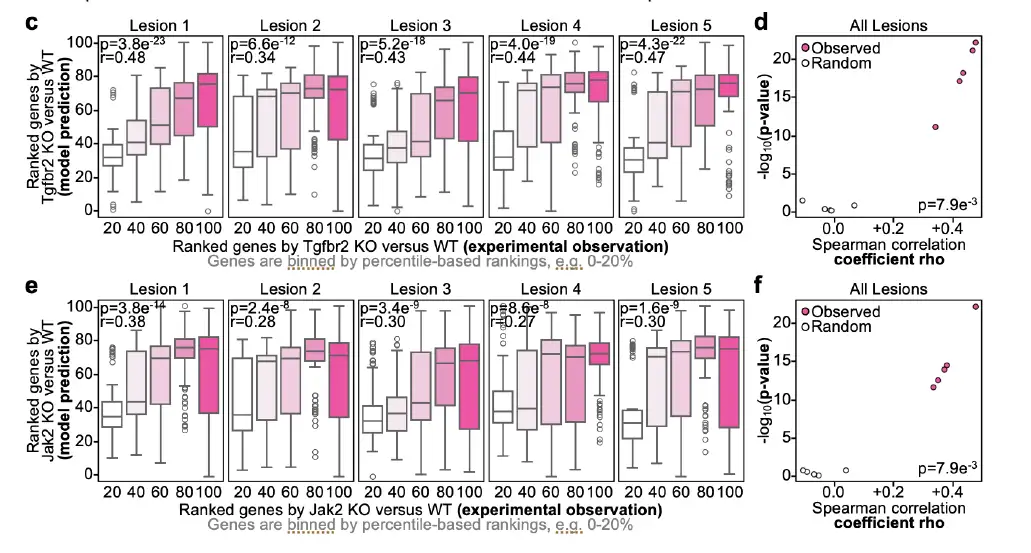
To demonstrate the effectiveness of Celcomen in correctly capturing perturbation effects in a spatial context, the researchers benchmarked it on an in vivo whole transcriptome dataset that measures gene knockdown in spatial transcriptomics.It's called Perturbmap. The Perturbmap dataset contains a mouse model for studying KP lung cancer, and in addition, there may be Jak2 or Tgfbr2 gene knockouts. The dataset annotates 5 spatial regions as lesion areas, and these regions are part of 1) KP wild-type cancer, or 2) KP cancer with Jak2 knockout, or 3) KP cancer with Tgfbr2 knockout, as shown below:
In the process of evaluating Celcomen's capabilities,The fetal spleen dataset used by the researchers comes from https://developmental.cellatlas.io/fetalimmune,Provided in log-normalized form, it is clear that log transformation and library size normalization were performed;Glioblastoma dataset from 10x Genomics,The same library size normalization, counts per million (CPM), and logarithmic transformation to base e were performed; in addition, only genes expressed in at least 100 cells were retained.
Model architecture: A new causal analysis framework Celcomen
The Celcomen model proposed in this study achieves causal inference identifiability and higher model interpretability by combining Lagrangian mechanics and causal inference. In simple terms, identifiability means whether the model can clearly identify causal relationships given sufficient data and reasonable assumptions, rather than obtaining the same observation results due to multiple different assumptions or model settings - this provides a new causal analysis framework for spatial transcriptomics research.
Celcomen is built on three core assumptions: ① The expected gene-gene correlation between first-order neighbors must exactly match the observed data; ② The expected gene-gene correlation within the same spatial point/cell must exactly match the observed data; ③ Causal Sufficiency Assumption: There is no unmeasured common cause between the gene pairs studied.
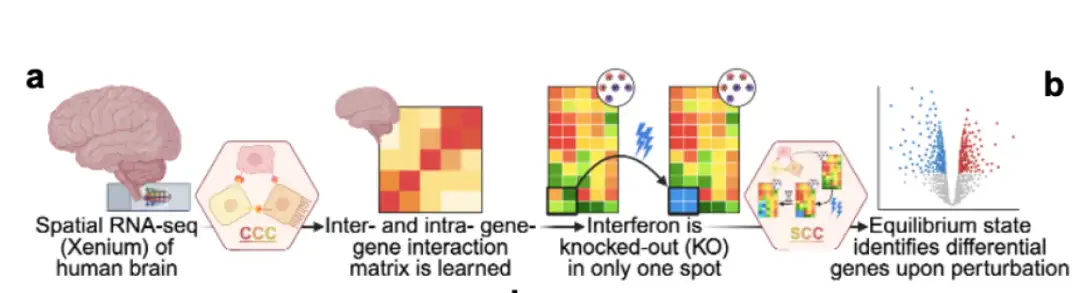
As shown in the following figure:Celcomen is divided into two parts: the reasoning module (CCE) and the generation module (SCE):
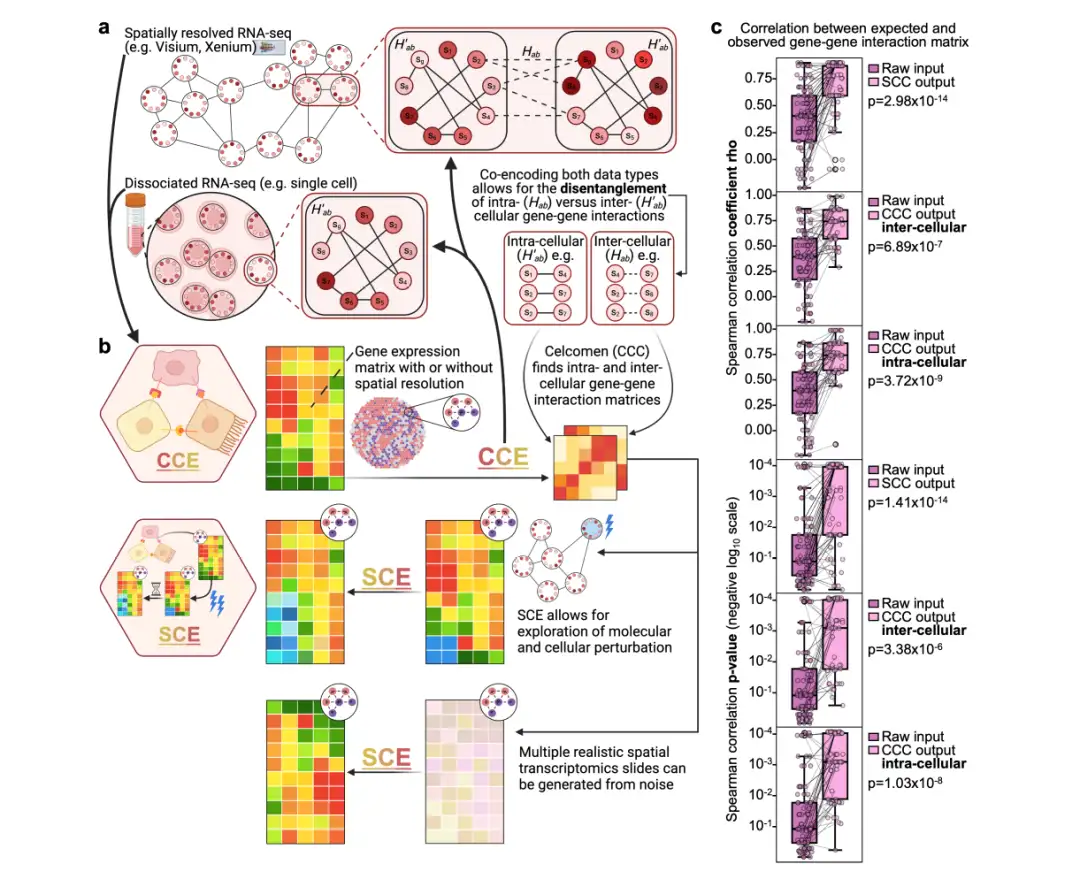
(a) Reasoning module (CCE):Gene-gene relationships can be learned from spatially resolved transcriptome data and optionally dissociated single-cell RNA-seq data. Highlighted cell-cell pairs in spatial data and individual cells in single-cell RNA-seq data demonstrate how CCE can distinguish intracellular (H′ab) from intercellular (Hab) gene-gene interactions.
(b) Generation module (SCE):Leverage gene-gene relationships learned by CCE to simulate counterfactual tissue behavior after cellular or genetic perturbations.
* Counterfactual scenario: This is a method used to study the possible behavior of biological tissues under different hypothetical conditions, mainly used for causal inference, intervention simulation and biomedical modeling. It involves constructing a hypothetical scenario, that is, if a key factor changes (such as gene knockout, drug intervention, external environment change, etc.), how the behavior of biological tissues will be different from the actual observed situation.
Research findings: Celcomen model is identifiable in disentangling causal relationships
The researchers verified the identifiability of the Celcomen model in learning causal structures and disentangling causal relationships through experiments on self-consistent synthetic data and real-world data.
Celcomen has strong self-consistency and identifiability
As shown in the figure below, on the synthetic dataset, Celcomen consistently shows strong consistency between its inferred gene-gene interactions and the real data, indicating that Celcomen has strong self-consistency and therefore identifiability.
* Self-consistency: In statistics, optimization, and machine learning, self-consistency usually means that the model's assumptions, derivations, and optimization processes can converge to a stable solution.
* Identifiability: refers to whether the model parameters or causal effects of the causal relationship can be uniquely determined based on the observed data in the causal inference model.
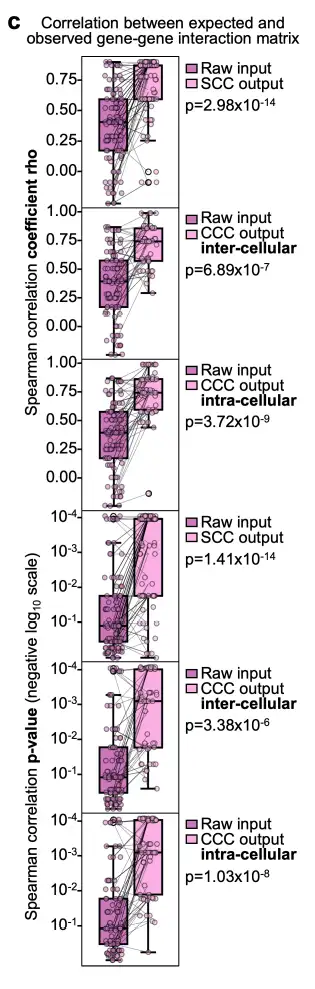
The researchers also confirmed the Celcomen model's identifiability guarantees on real human data by applying it to spatial transcriptome sections of multiple human fetal spleens, and observed Spearman correlation coefficients between the two gene-gene interaction matrices in the range of 0.5-0.6. Furthermore, the captured gene interactions in both intra- and inter-cellular matrices were biologically plausible as they followed known biological intra- and inter-cellular processes.
This demonstrates the identifiability of Celcomen, confirming its implied stability and robustness beyond theoretical and synthetic data and also observable in real human samples.
Causal decoupling capability: Celcomen can successfully disentangle the sources of intrinsic and extrinsic transcriptome variation
Next, the researchers tested Celcomen's ability to untangle the gene regulation programs within and between cells (decoupling ability). They applied Celcomen in a real human clinical setting to analyze a single-cell resolution spatial transcriptome dataset of human glioblastoma (brain cancer). As shown in the figure below, the researchers found that Celcomen was able to successfully untangle the sources of intrinsic and extrinsic transcriptome variation.
In vivo spatial counterfactual validation: Celcomen performs significantly better than a random baseline
To further demonstrate the effectiveness of Celcomen, the researchers benchmarked it on the in vivo whole transcriptome dataset Perturbmap. The results showed that for all lesions, the Spearman correlation between the predictions and the in vivo measurements ranged from 0.28-0.47. To evaluate the significance of this performance, the researchers compared the model to a random benchmark, where Celcomen was run on randomly shuffled data. The results showed that Celcomen performed significantly better than the random benchmark, with a p-value of 0.0079, as shown in the figure below (cf):
In summary, the model proposed in the study opens a new path for achieving mechanistic interpretability through causal inference. As demonstrated in the experiment, due to the causal identifiability of the Celcomen model, researchers can recover the parameter values of the neural network with high accuracy. Celcomen's progress has a significant impact on the biomedical field, such as by revealing how diseases lead to tissue failure and promoting testable hypotheses about treatment benefits. As the technology advances, Celcomen's value will continue to grow, promoting the improvement of disease modeling and mechanistic understanding.
Artificial intelligence unlocks the potential of spatial transcriptomics
The relevant results achieved in this study are another development of spatial transcriptomics - spatial transcriptomics technology is one of the major breakthroughs in the field of bioinformatics in recent years. This technology has greatly changed the paradigm of biomedical research by providing detailed, spatially located molecular features, enabling biological researchers to elucidate tissue structure and function with unprecedented resolution.
In the past few years, spatial transcriptomics technology has achieved rapid development and data has been continuously accumulated. On this basis, the article "Nature Methods Special Issue Comment: Using the "Key" of Artificial Intelligence to Open the "Lock" of Spatial Omics" published in August 2024 pointed out thatArtificial intelligence has the potential to unlock the full potential of spatial omics, facilitating the integration of complex datasets and discovering new biomedical insights.
Specifically, AI can facilitate the integration of spatial transcriptomics and scRNA-seq, allowing researchers to measure spatial gene expression profiles across the transcriptome at the single-cell level. In addition, by integrating spatial omics with histological imaging data, AI can construct high-resolution, comprehensive three-dimensional spatial tissue maps covering a wide range of omics modalities. As the number of available datasets grows, multimodal large language models (MM-LLMs) can be trained on spatial omics, medical imaging, and clinical text data for tasks in biomedical research and precision medicine.
October 2023Zhang Shihua's research group at the Institute of Mathematics and Systems Science, Chinese Academy of Sciences, published a paper in Nature Computational Science.Published a research paper titled "Integrating spatial transcriptomics data across different conditions, technologies, and developmental stages". This work established a new integrated analysis tool STAligner for spatial transcriptomics data from multiple slices of biological tissues from different technologies, different developmental time points, and different disease conditions, which can help researchers discover new important biological insights when conducting spatial transcriptomics analysis.
*Original paper:
https://www.biorxiv.org/content/10.1101/2022.12.26.521888v1.full.pdf
In order to solve the multi-faceted challenges faced by spatial transcriptome data analysis, in July 2024,Associate Professor Zhang Qiangfeng's research group at the School of Life Sciences, Tsinghua University/Structural Biology Advanced Innovation Center/Tsinghua-Peking University Joint Center for Life Sciences,A research paper titled "Tissue module discovery in single-cell resolution spatial transcriptomics data via cell-cell interaction-aware cell embedding" was published online in the journal Cell Systems. The study developed an artificial intelligence algorithm SPACE (spatial transcriptomics data analysis via "interaction-aware" cell embedding) based on the graph autoencoder deep learning framework, which can identify spatial cell types and discover tissue modules from spatial transcriptome data with single-cell resolution, and can be used for large-scale spatial transcriptome research.
Looking ahead, by leveraging the powerful computing power and deep learning algorithms of AI, researchers are expected to unlock a new dimension of spatial transcriptomics, significantly improve the efficiency of disease research, drug development, and personalized medicine, and enable scientists to explore the spatial heterogeneity of biological systems with unprecedented precision, thereby bringing groundbreaking scientific discoveries.
References:
1.https://openreview.net/forum?id=Tqdsruwyac
2.https://www.thepaper.cn/newsDetail_forward_28521641
3.https://www.cas.cn/syky/202310/t20231020_4981872.shtml








