Command Palette
Search for a command to run...
LAMMPS: Am Beispiel Von Einkristall-Aluminium Wird Die Einachsige Spannung Von Materialien Simuliert
LAMMPS (Large-scale Atomic/Molecular Massively Parallel Simulator) ist ein klassischer Molekulardynamik-Simulationscode mit Schwerpunkt auf der Materialmodellierung. Er ist für den effizienten Betrieb auf Parallelrechnern sowie eine einfache Erweiterung und Modifikation konzipiert. LAMMPS wurde ursprünglich von den Sandia National Laboratories, einer Behörde des US-Energieministeriums, entwickelt und umfasst heute Beiträge zahlreicher Forschungsgruppen und Einzelpersonen aus zahlreichen Institutionen. Der Großteil der Finanzierung von LAMMPS stammt vom US-Energieministerium. LAMMPS ist Open-Source-Software und wird unter den Bedingungen der GNU Public License Version 2 (GPLv2) vertrieben.
1. Tutorial-Inhalte
In diesem Tutorial simulieren wir die einachsige Dehnung des Materials durch Änderung der Gitterkonstante. Anschließend berechnen und zeichnen wir die Dehnungs-Spannungs-Kurve des Materials. In diesem Tutorial lernen Sie:
- Verwenden Sie npt, um die Systemstruktur zu entspannen
- Verwenden Sie fix in Verbindung mit dem Variablenbefehl, um die Gitterkonstante zu ändern
- Verwenden Sie den Compute-Befehl, um die Systembelastung zu berechnen

2. Eingabedatei
- Al99.Eam.Legierung EAM-Potenzial von Werkstoffen
- in.txt Eingabedatei
- pl.py Bildskript zeichnen
3. Analyse der Eingabedatei
Systeminitialisierung und Grundeinstellungen
units metal # 采用金属单位制(长度:Å,时间:ps,质量:g/mol,能量:eV 等)
dimension 3 # 三维空间模拟
boundary p p p # 三个方向均为周期性边界条件(模拟无限大晶体)
atom_style atomic # 原子类型为「基本原子」,仅包含位置和类型信息
variable latparam equal 4.05 # 定义铝的晶格常数为 4.05Å(FCC 铝的典型值)
Aufbau der Atomstruktur
lattice fcc ${latparam} orient x 1 0 0 orient y 0 1 0 orient z 0 0 1 # 定义 FCC 晶格 # 晶格常数为 ${latparam},晶向沿 x[100] 、 y[010] 、 z[001](标准笛卡尔坐标系)region whole block 0 10 0 10 0 10 # 定义模拟区域:一个立方体,在 x 、 y 、 z 方向各包含 10 个晶格常数长度
create_box 1 whole # 创建容纳原子的「盒子」,仅包含 1 种原子类型,边界为上述 region 定义的范围
create_atoms 1 region whole # 在盒子内填充类型为 1 的原子,按 FCC 晶格排列
Einstellungen des interatomaren Wechselwirkungspotentials
pair_style eam/alloy # 采用嵌入原子法(EAM)合金势描述原子间相互作用(适合金属体系)
pair_coeff * * Al99.eam.alloy Al # 配置势函数参数:
# - 第一个*:所有原子类型(此处仅 1 种)
# - 第二个*:所有原子类型的相互作用
# - Al99.eam.alloy:势函数文件(预训练的铝势)
# - Al:指定原子类型 1 对应元素为铝
Definition des Berechnungsvolumens (für spätere Analysen)
compute csym all centro/atom fcc # 计算每个原子的中心对称参数(用于识别 FCC 结构完整性,缺陷处会偏离)
compute peratom all pe/atom # 计算每个原子的势能(用于分析能量分布)
Gleichgewichtssimulationsphase (Beseitigung der anfänglichen Spannung und Erreichen eines stabilen Zustands)
reset_timestep 0 # 重置时间步计数器为 0(平衡阶段作为新起点) timestep 0.001 # 时间步长为 0.001ps(即 1fs,金属模拟常用精度) #初始化原子速度:温度 300K(室温),随机种子 12345,消除整体动量(mom yes)和旋转(rot no)
velocity all create 300 12345 mom yes rot no #施加 NPT 系综(恒温、恒压、恒粒子数)进行平衡:
#- temp 300 300 1:温度维持 300K,热浴阻尼系数 1(单位:ps)
#- iso 0 0 1:各向同性压力控制,目标压力 0bar,压力阻尼系数 1(单位:ps)
#- drag 1:原子运动阻尼系数(用于稳定系综)
fix 1 all npt temp 300 300 1 iso 0 0 1 drag 1 #热力学输出设置:
thermo 1000 #每 1000 步输出一次热力学信息
thermo_style custom step lx ly lz press pxx pyy pzz pe temp #输出内容:
#步数、盒子尺寸(x/y/z)、总压力、压力张量分量、势能、温度 run 20000 #运行 20000 步平衡模拟(总时间 20000×0.001ps=20ps)
unfix 1 #平衡结束,移除 NPT 约束#记录平衡后的 x 方向盒子长度(用于后续计算应变)
variable tmp equal "lx" #临时变量存储当前 x 方向长度
variable L0 equal ${tmp} #定义 L0 为初始长度(拉伸前的平衡长度)
print "Initial Length, L0: ${L0}" #输出初始长度,便于验证
Einachsige Zugverformungsstufe
reset_timestep 0 #重置时间步计数器为 0(拉伸阶段作为新起点) #重新施加 NPT 系综,但仅控制温度和横向压力:
#- temp 300 300 1:维持温度 300K
#- y 0 0 1 z 0 0 1:y 和 z 方向压力保持 0bar(横向自由,模拟单轴拉伸)
fix 1 all npt temp 300 300 1 y 0 0 1 z 0 0 1 drag 1 #定义拉伸速率:
variable srate equal 1.0e10 #应变速率为 1e10 /s(分子动力学常用高应变速率,远高于实验)
variable srate1 equal "v_srate / 1.0e12" #转换为 LAMMPS 单位(应变速率单位:ps⁻¹,1e10/s = 1e-2 ps⁻¹)#施加 x 方向拉伸变形:
#- deform 1 x:沿 x 方向变形,变形组为 1(所有原子)
#- erate ${srate1}:按上述应变速率拉伸
#- units box:变形基于盒子尺寸
#- remap x:原子坐标随盒子拉伸同步更新(避免原子「跑出」盒子)
fix 2 all deform 1 x erate ${srate1} units box remap x
Stretching der Prozessdatenausgabe
#定义应变和应力变量: variable strain equal "(lx - v_L0)/v_L0" #工程应变 =(当前 x 长度 - 初始长度)/ 初始长度 variable p1 equal "v_strain" #p1 对应工程应变(无量纲) variable p2 equal "-pxx/10000" #x 方向应力(GPa):pxx 为 LAMMPS 内置压力张量(单位 bar),负号转为拉应力,除以 10000 转换为 GPa variable p3 equal "-pyy/10000" #y 方向应力(GPa) variable p4 equal "-pzz/10000" #z 方向应力(GPa) #输出应变-应力数据到文件:
#- 每 100 步输出一次 p1(应变)、 p2(x 应力)、 p3(y 应力)、 p4(z 应力)
#- 保存到 Al_tens_100.def1.txt,不在屏幕显示
fix def1 all print 100 "${p1} ${p2} ${p3} ${p4}" file Al_tens_100.def1.txt screen no #输出原子轨迹文件(用于可视化):
#- 每 100 步保存一次,文件名为 md.lammpstrj
#- 包含原子 ID 、类型、 x/y/z 坐标
dump trace all custom 100 md.lammpstrj id type x y z #热力学信息输出设置:
thermo 1000 #每 1000 步输出一次
thermo_style custom step v_strain temp v_p2 v_p3 v_p4 ke pe press #输出步数、应变、温度、三方向应力、动能、势能、总压力run 30000 #拉伸阶段运行 30000 步(总时间 30ps,累积应变约 0.3)
Ende der Simulation
print "All done" # 输出完成信息,提示模拟结束
4. Erste Schritte mit der Simulation
1. 克隆本容器后进入容器设置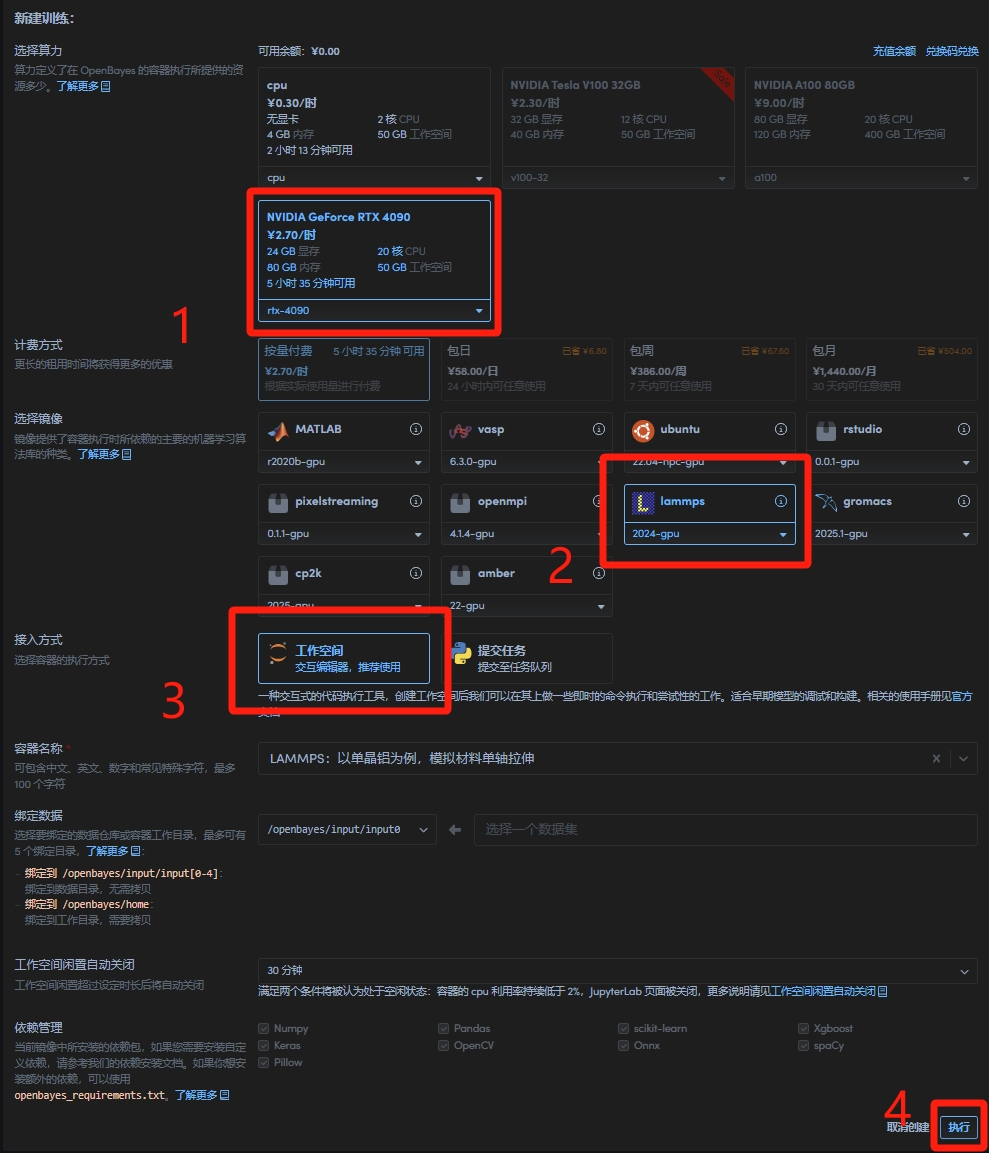
2. 待加载完毕后打开工作空间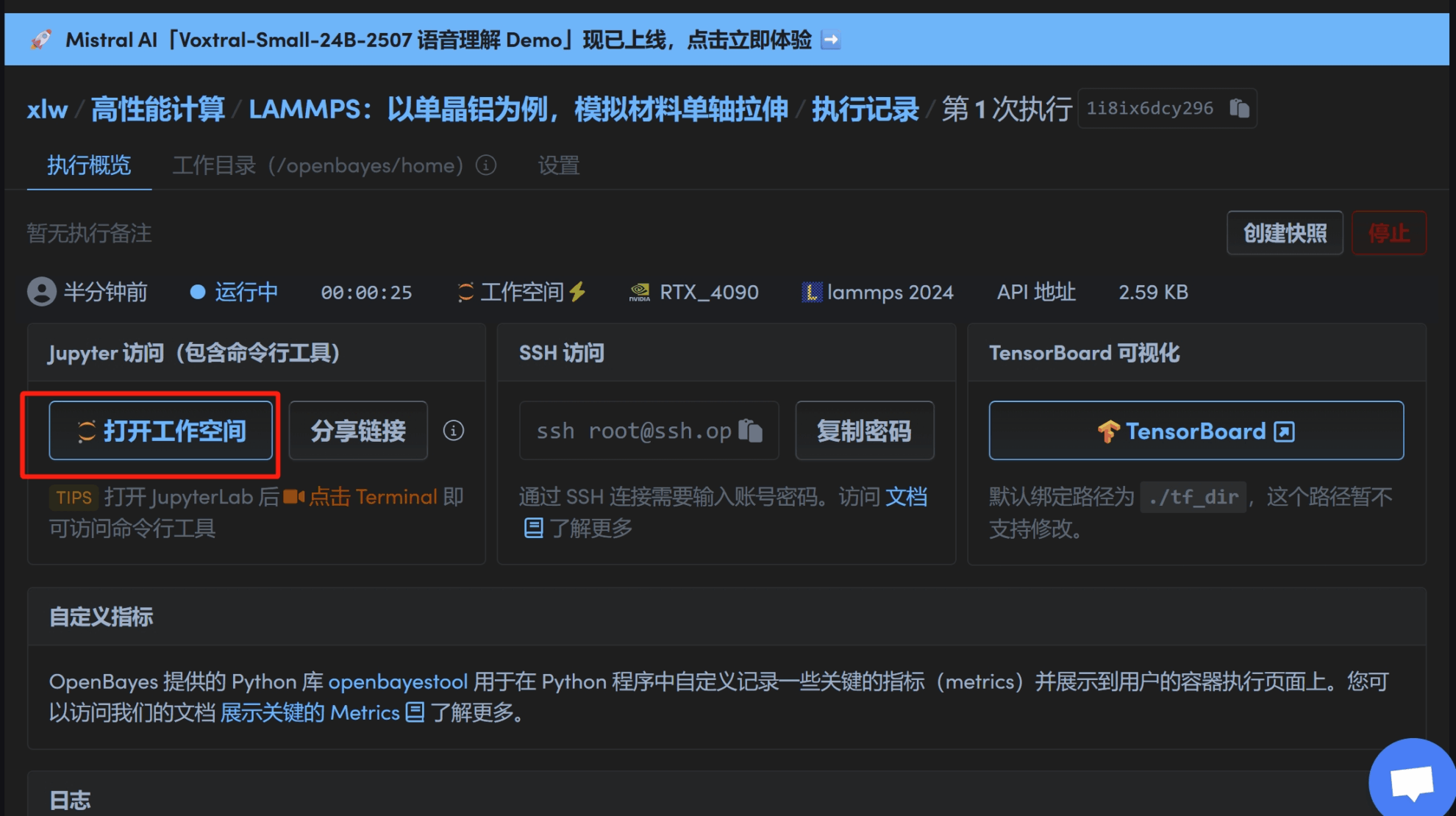
3. 随后打开终端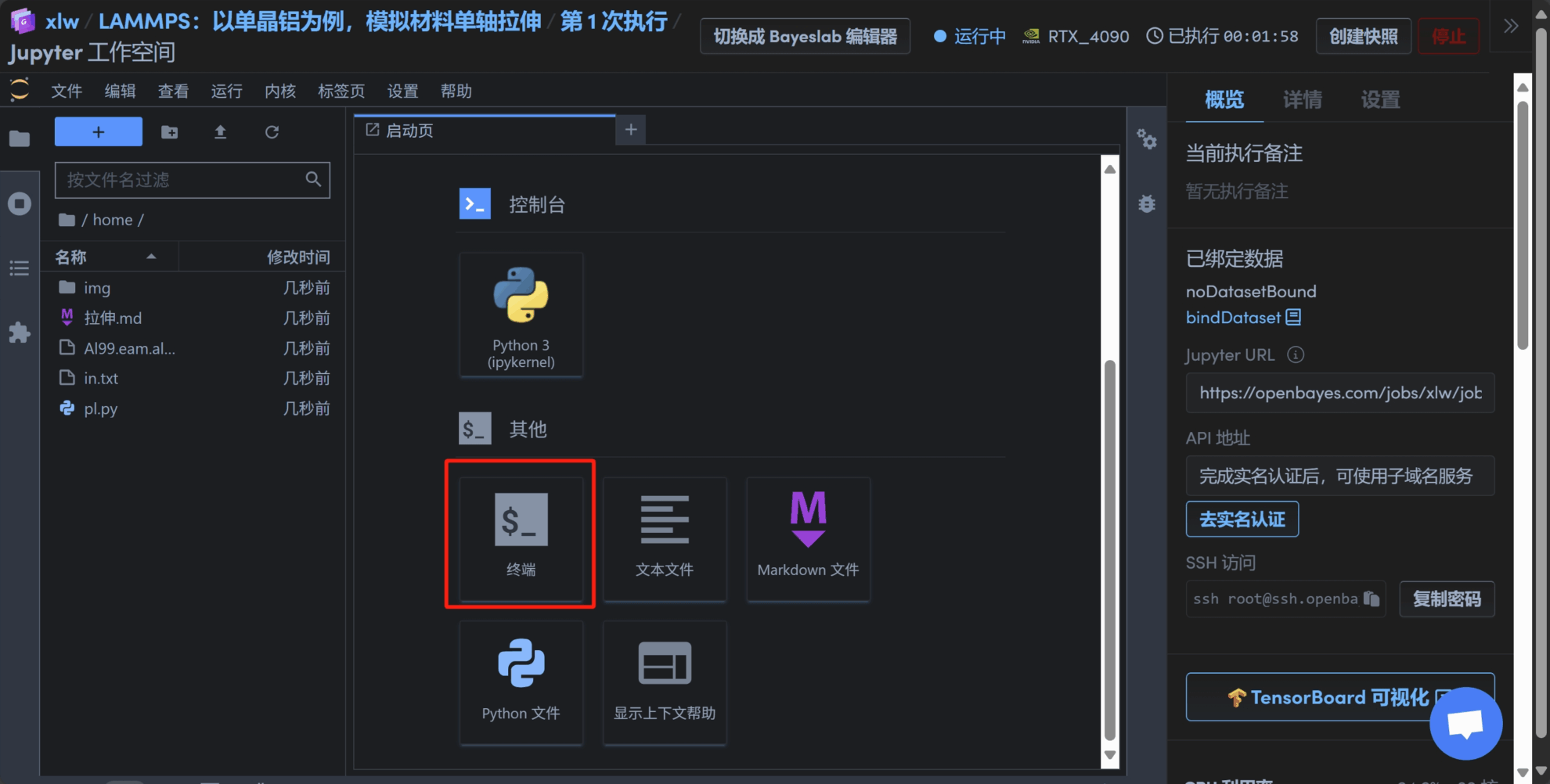
4. 输入以下命令运行 lammpsmpirun --allow-run-as-root -np 2 lmp -sf gpu -pk gpu 1 < in.txt 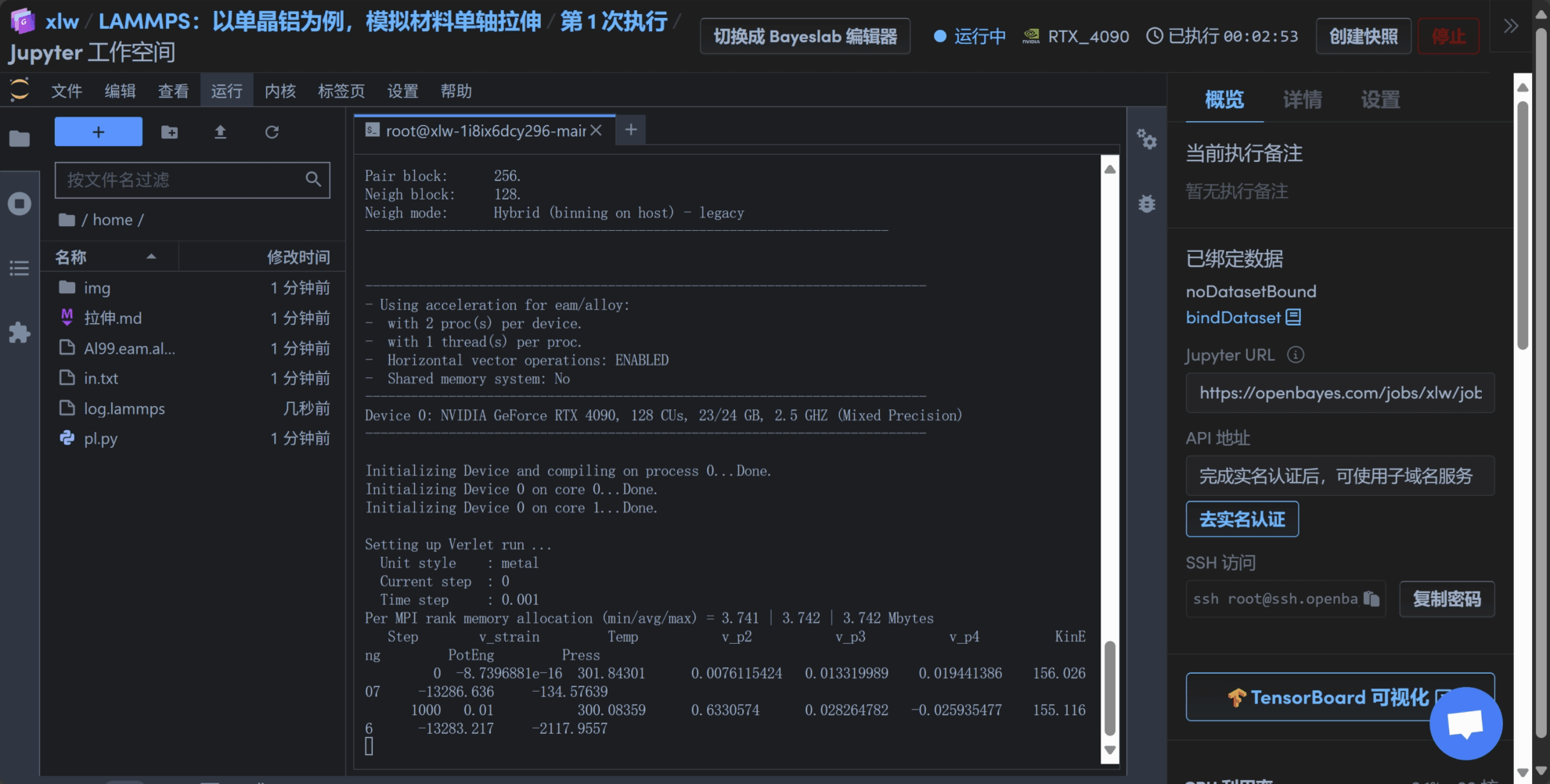
5. Dateiausgabe
- Al_tens_100.def1.txt:Daten aus Dehnungs-Spannungs-Kurven können zur Analyse mechanischer Eigenschaften wie Elastizitätsmodul und Streckgrenze verwendet werden.
- md.lammpstrj: Atomtrajektoriendateien, die zur Visualisierung der Kristallstrukturentwicklung während der Dehnung durch Software wie VMD und Ovito verwendet werden können
6. Zeichnen von Dehnungs-Spannungs-Diagrammen
1. 在终端输入以下命令python pl.py2. 打开绘制的图片
3. 可以得到曲线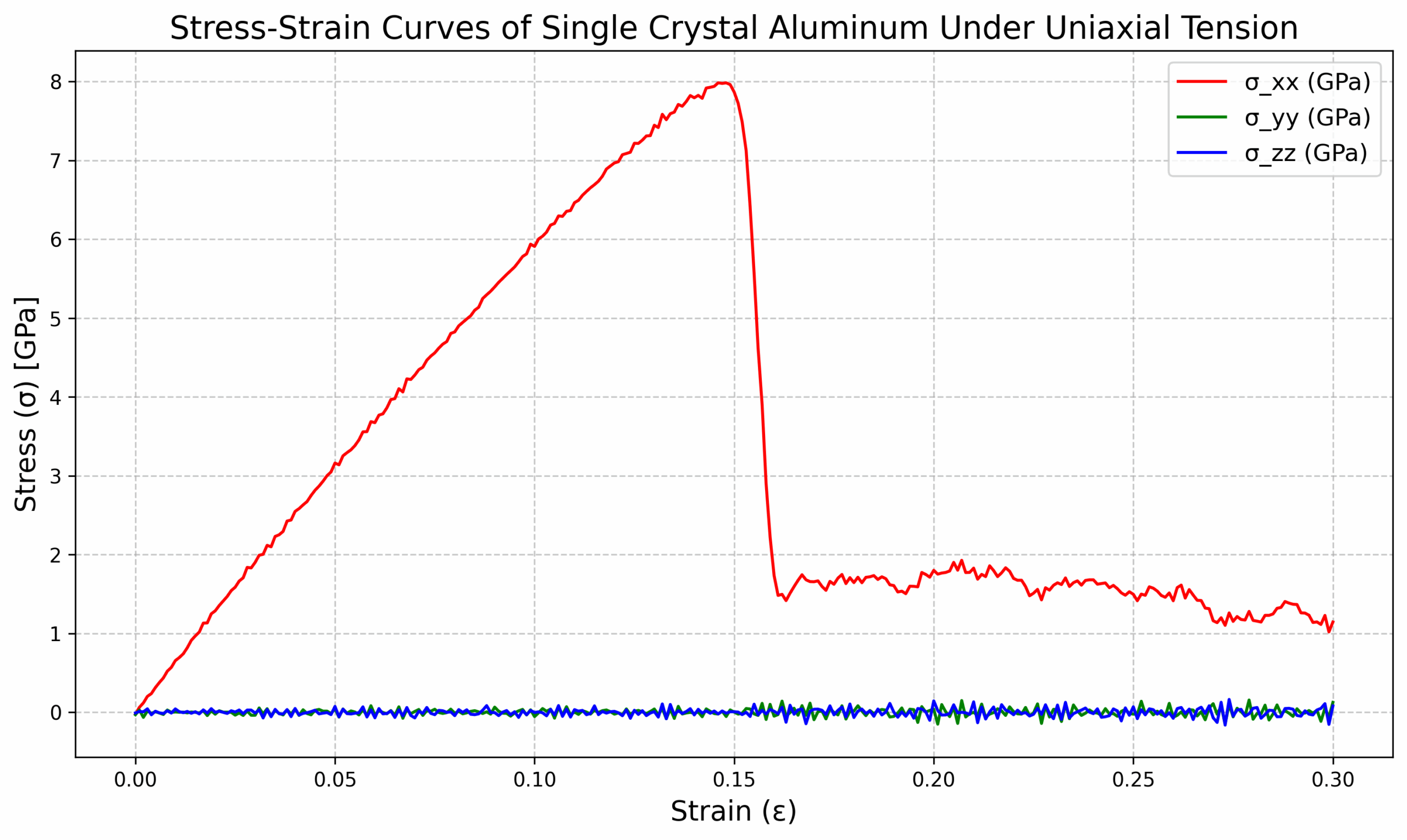
KI mit KI entwickeln
Von der Idee bis zum Launch – beschleunigen Sie Ihre KI-Entwicklung mit kostenlosem KI-Co-Coding, sofort einsatzbereiter Umgebung und bestem GPU-Preis.