Command Palette
Search for a command to run...
Präzise Vorhersage neuartiger biomolekularer Interaktionen mit IsoDDE
Präzise Vorhersage neuartiger biomolekularer Interaktionen mit IsoDDE
Isomorphic Labs Team
Zusammenfassung
Die Vorhersage biomolekularer Interaktionen ist eine grundlegende Voraussetzung für das rationale Wirkstoffdesign (rational drug design). Dennoch bleibt die Erzielung experimenteller Genauigkeit und Generalisierbarkeit über neuartige chemische Räume hinweg ein kritischer Engpass. Während Deep-Learning-Ansätze wie AlphaFold 3 die Strukturvorhersage vorangebracht haben, zeigen Benchmarks, dass weiterhin Einschränkungen bei der Generalisierung auf unerschlossene Regionen des Molekülraums, der Schätzung der Bindungsaffinität sowie der Detektion molekularer Bindungsstellen auf bisher nicht charakterisierten Proteinoberflächen bestehen.In dieser Arbeit stellen wir die Isomorphic Labs Drug Design Engine (IsoDDE) vor, ein einheitliches computergestütztes System, das darauf ausgelegt ist, diese Limitationen zu adressieren. Wir demonstrieren, dass IsoDDE die Genauigkeit von AlphaFold 3 bei einem anspruchsvollen Benchmark zur Protein-Ligand-Generalisierung mehr als verdoppelt, indem es komplexe Out-of-Distribution-Ereignisse wie Induced Fits erfolgreich modelliert und neuartige Bindungstaschen (binding pockets) präzise identifiziert. Im Bereich der Biologika übertrifft IsoDDE bestehende Modelle erheblich und setzt einen neuen State-of-the-Art für die Vorhersage von Antikörper-Antigen-Schnittstellen sowie die Modellierung der CDR-H3-Schleife.Schließlich übertreffen die Affinitätsprognosen von IsoDDE für niedermolekulare Binder (small molecule binders) die Goldstandard-Methoden der physikbasierten Modellierung. Damit schließt das System die Lücke hin zu experimenteller Präzision, ohne den Rechenaufwand traditioneller physikbasierter Workflows in Kauf nehmen zu müssen. Unsere Ergebnisse zeigen, dass IsoDDE eine skalierbare Grundlage für das KI-gestützte Wirkstoffdesign bietet und die prädiktive Treue (predictive fidelity) bereitstellt, die erforderlich ist, um neuartige biologische Systeme mit einer beispiellosen Genauigkeit zu explorieren.
One-sentence Summary
The Isomorphic Labs Drug Design Engine (IsoDDE) is a unified computational system that outperforms AlphaFold 3 on protein-ligand generalization benchmarks by modeling complex out-of-distribution events like induced fits, achieving state-of-the-art accuracy in antibody-antigen interface and CDR-H3 loop modeling, and providing small molecule binding affinity predictions that exceed gold-standard physics-based methods.
Key Contributions
- The paper introduces the Isomorphic Labs Drug Design Engine (IsoDDE), a unified computational system that achieves a step change in unconditional accuracy for biomolecular interaction prediction. This architecture demonstrates the ability to more than double the accuracy of AlphaFold 3 on protein-ligand generalization benchmarks and successfully models complex out-of-distribution events like induced fits.
- This work presents a high-performance model for the biologics domain that establishes a new state of the art in antibody-antigen interface prediction and CDR-H3 loop modeling. The system also enables blind pocket identification, allowing for the discovery of novel binding sites on previously uncharacterized protein surfaces without requiring a specified ligand.
- The research provides a quantitative binding affinity estimation capability that outperforms gold-standard physics-based methods for small molecule binders. This approach bridges the gap toward experimental-grade precision while avoiding the high computational overhead associated with traditional free energy perturbation workflows.
Introduction
Accurate prediction of biomolecular interactions is essential for rational drug design and the discovery of new biological mechanisms. While deep learning models like AlphaFold 3 have advanced structure prediction, they often struggle to generalize to unexplored chemical spaces, fail to identify novel binding pockets, and lack the ability to provide precise quantitative binding affinity. Current methods for estimating affinity are either computationally expensive physics-based simulations or deep learning models that correlate poorly with experimental data. The authors introduce the Isomorphic Labs Drug Design Engine (IsoDDE), a unified computational system that achieves a step change in accuracy and generalization. IsoDDE more than doubles the accuracy of previous state-of-the-art models on protein-ligand benchmarks, provides superior antibody-antigen interface modeling, and delivers binding affinity predictions that exceed gold-standard physics-based methods.
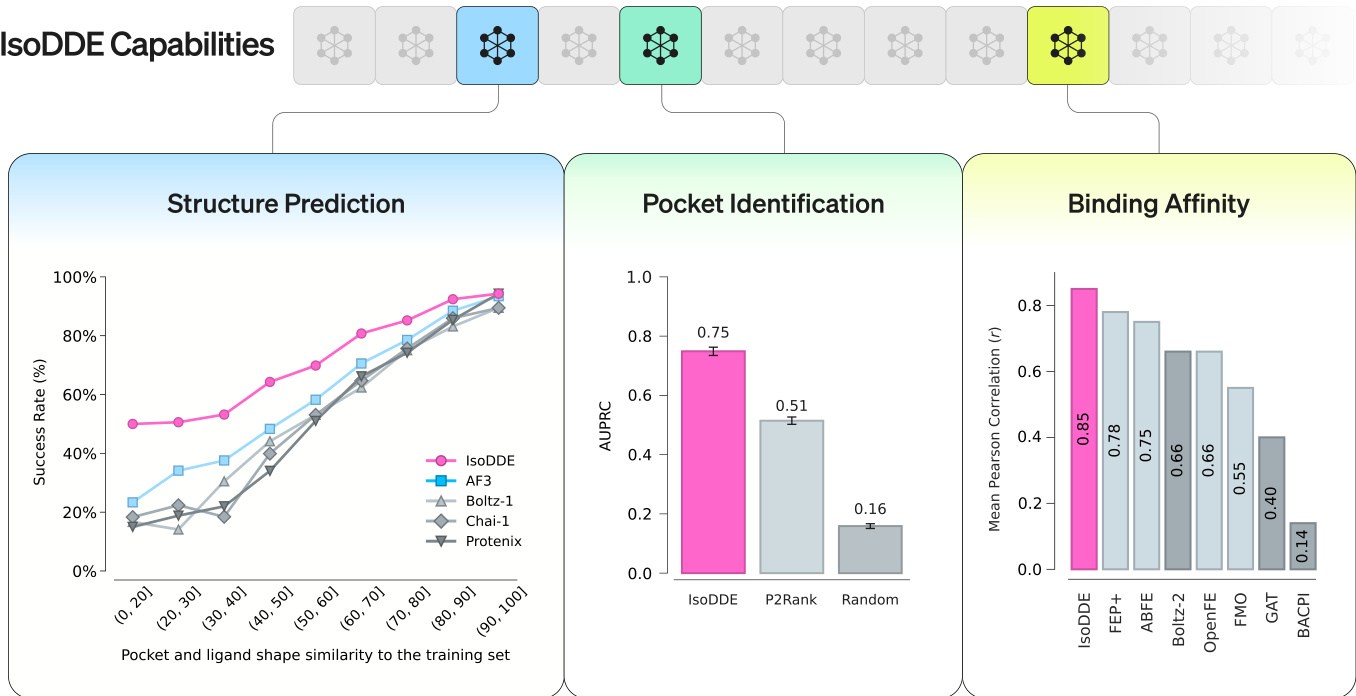
Experiment
IsoDDE was evaluated across multiple benchmarks, including Runs N' Poses, FoldBench, and antibody-antigen datasets, to assess its structural modeling and generalization capabilities. The results demonstrate that the model significantly outperforms previous state-of-the-art methods like AlphaFold 3 and Boltz-2, particularly when predicting highly novel protein-ligand interfaces and complex antibody-antigen structures. Furthermore, IsoDDE shows exceptional performance in predicting binding affinities and identifying cryptic pockets, proving to be a robust tool for navigating diverse chemical and biological spaces in drug discovery.
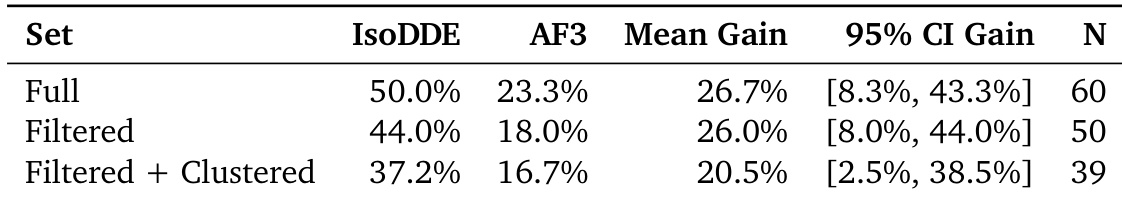
The authors compare the performance of IsoDDE against AF3 across different subsets of the Runs N' Poses benchmark. Results show that IsoDDE achieves a higher success rate than AF3 in the full dataset, the filtered dataset, and the filtered plus clustered dataset. IsoDDE demonstrates a consistent performance advantage over AF3 across all evaluated data subsets The mean gain in success rate remains substantial whether using the full or filtered datasets IsoDDE maintains superior performance even when the data is filtered and clustered to reduce redundancy
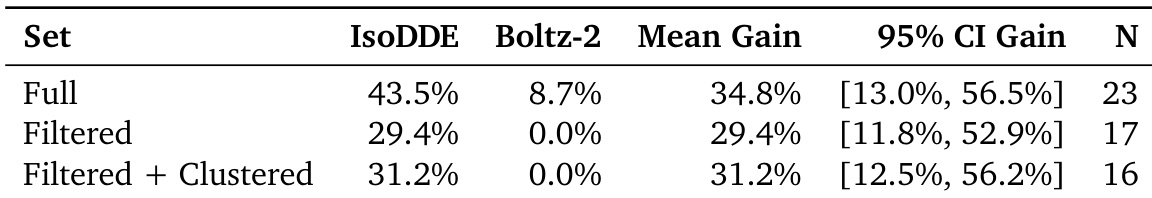
The authors evaluate the performance of IsoDDE against Boltz-2 across different data subsets. Results show that IsoDDE maintains a higher success rate than Boltz-2 in the full, filtered, and clustered datasets. IsoDDE demonstrates a substantial mean gain in performance compared to Boltz-2 across all tested sets The model maintains consistent improvements even when data is filtered or clustered Performance advantages are observed in both the full dataset and more specialized subsets
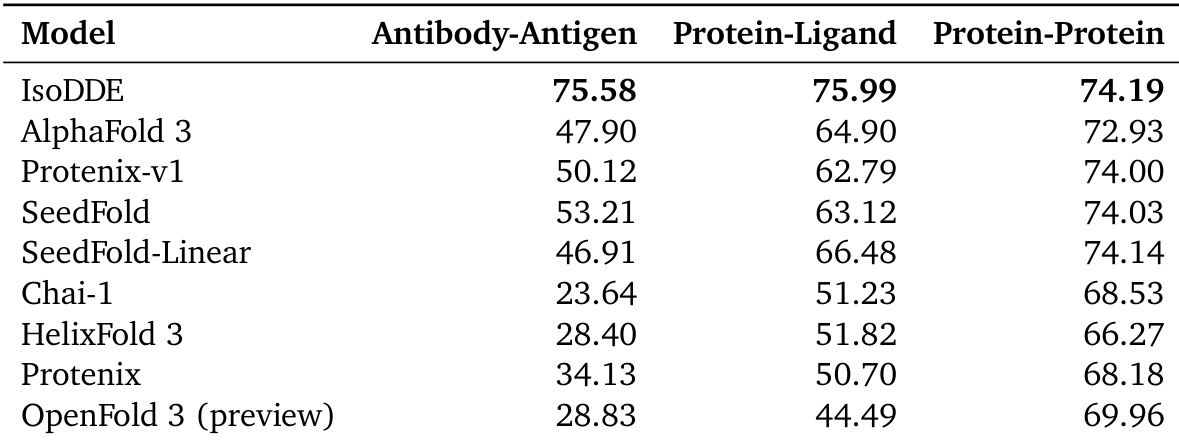
The authors evaluate IsoDDE's structural modeling performance on the FoldBench dataset across three distinct interface types. Results show that IsoDDE achieves superior success rates compared to several external models in antibody-antigen, protein-ligand, and protein-protein benchmarks. IsoDDE demonstrates higher success rates in antibody-antigen interface prediction than all compared models The model shows improved performance in protein-ligand modeling relative to the other evaluated methods IsoDDE maintains a competitive advantage across protein-protein interaction benchmarks
IsoDDE was evaluated against AF3, Boltz-2, and various external models using the Runs N' Poses and FoldBench benchmarks to assess its structural modeling capabilities. Across diverse datasets and interface types, including antibody-antigen, protein-ligand, and protein-protein interactions, IsoDDE consistently demonstrated superior success rates. The results indicate that the model maintains a robust performance advantage even when data is filtered or clustered to reduce redundancy.