Command Palette
Search for a command to run...
dnaHNet: Ein skalierbares und hierarchisches Foundation Model für das Lernen genomischer Sequenzen
dnaHNet: Ein skalierbares und hierarchisches Foundation Model für das Lernen genomischer Sequenzen
Arnav Shah Junzhe Li Parsa Idehpour Adibvafa Fallahpour Brandon Wang Sukjun Hwang Bo Wang Patrick D. Hsu Hani Goodarzi Albert Gu
Zusammenfassung
Genomische Foundation-Modelle besitzen das Potenzial, die Syntax der DNA zu entschlüsseln, stehen jedoch vor einem grundlegenden Kompromiss bei ihrer Input-Repräsentation. Standardmäßige Tokenizer mit festem Vokabular fragmentieren biologisch bedeutsame Motive wie Codons und regulatorische Elemente, während Modelle auf Nukleotid-Ebene die biologische Kohärenz bewahren, jedoch prohibitiv hohe Rechenkosten bei langen Kontexten verursachen.Wir führen dnaHNet ein, ein hochmodernes, tokenizer-freies autoregressives Modell, das genomische Sequenzen end-to-end segmentiert und modelliert. Durch einen differenzierbaren Mechanismus zum dynamischen Chunking (differentiable dynamic chunking mechanism) komprimiert dnaHNet rohe Nukleotide adaptiv in latente Tokens und balanciert dabei die Kompression mit der Vorhersagegenauigkeit. Prätrainiert auf prokaryotischen Genomen, übertrifft dnaHNet führende Architekturen, einschließlich StripedHyena2, in Bezug auf Skalierbarkeit und Effizienz. Dieses rekursive Chunking führt zu einer quadratischen Reduktion der FLOPs, was eine mehr als 3-fache Beschleunigung der Inferenz gegenüber Transformers ermöglicht.Bei Zero-Shot-Aufgaben erzielt dnaHNet eine überlegene Leistung bei der Vorhersage der Fitness von Proteinvarianten sowie der Gen-Essentialität, während es gleichzeitig hierarchische biologische Strukturen ohne Supervision automatisch entdeckt. Diese Ergebnisse etablieren dnaHNet als ein skalierbares und interpretierbares Framework für die genomische Modellierung der nächsten Generation.
One-sentence Summary
By employing a differentiable dynamic chunking mechanism to adaptively compress raw nucleotides into latent tokens, the tokenizer-free autoregressive foundation model dnaHNet achieves superior scaling and efficiency over architectures like StripedHyena2 and Transformers, delivering over 3x inference speedup and state-of-the-art performance on zero-shot tasks such as predicting protein variant fitness and gene essentiality.
Key Contributions
- The paper introduces dnaHNet, a tokenizer-free autoregressive model that utilizes a differentiable dynamic chunking mechanism to adaptively compress raw nucleotides into latent tokens.
- This architecture achieves significant computational efficiency by employing recursive chunking to reduce FLOPs quadratically, resulting in over 3x faster inference speeds compared to Transformer models.
- Experiments on prokaryotic genomes demonstrate that the model outperforms leading architectures like StripedHyena2 in scaling and efficiency, while also showing superior zero-shot performance in predicting protein variant fitness and gene essentiality.
Introduction
Genomic foundation models are essential for decoding DNA syntax to advance fields like drug discovery and synthetic biology. However, current approaches face a fundamental tradeoff between computational efficiency and biological accuracy. Fixed-vocabulary tokenizers are efficient but often fragment critical biological motifs like codons, while nucleotide-level models preserve biological coherence but suffer from prohibitive computational costs when processing long genomic contexts. The authors leverage a hierarchical, tokenizer-free architecture called dnaHNet to resolve this tension. By using a differentiable dynamic chunking mechanism, the model adaptively compresses raw nucleotides into latent tokens, allowing it to achieve superior scaling, faster inference, and state-of-the-art performance on zero-shot tasks such as protein variant fitness prediction.
Dataset
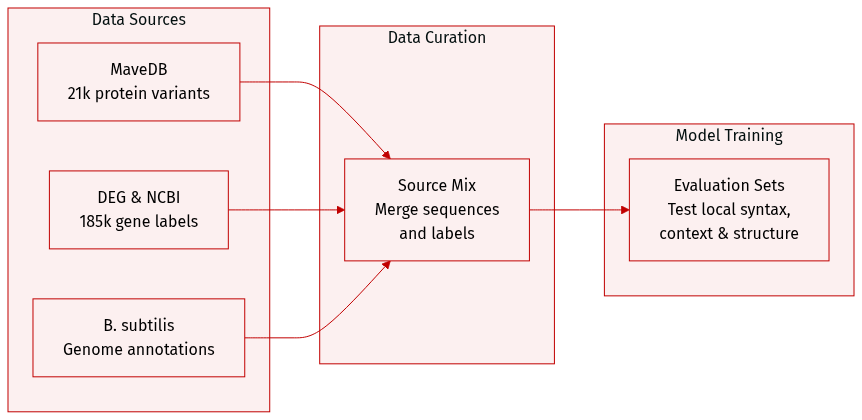
The authors evaluate dnaHNet using three distinct datasets designed to test different biological modeling capabilities:
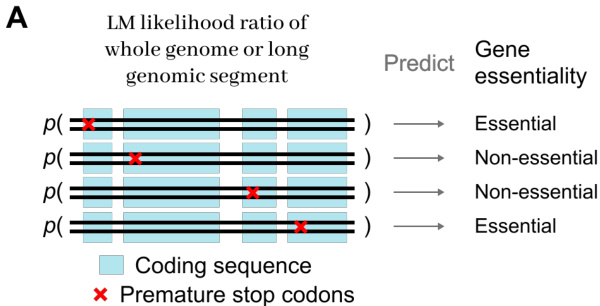
- Protein Variant Effects (MaveDB): This subset consists of 21,250 nucleotide-level data points compiled from 12 experimental fitness datasets for E. coli K-12. It is used to assess the model's ability to capture local coding syntax and predict protein fitness landscapes.
- Gene Essentiality (DEG): Comprising 185,226 data points, this dataset was constructed by generating binary essentiality labels for genes across 62 bacterial organisms from the Database of Essential Genes. The authors sourced base sequences and annotations from NCBI and labeled genes as essential if they matched DEG entries by name or sequence identity greater than 99 percent. This subset evaluates the model's capacity to integrate genomic context and long-range dependencies.
- Genomic Structure Interpretation (NCBI): To perform interpretability analysis, the authors used the B. subtilis genome and functional annotations from NCBI. They partitioned the genome into distinct functional regions based on these annotations to determine how the model's segmentation aligns with known biological structures.
Method
The authors introduce dnaHNet, a scalable, tokenizer-free foundation model designed for genomic sequence learning. The model formulates genomic learning as an autoregressive sequence modeling problem. Given a nucleotide sequence X=(x1,…,xL) where xt∈{A,C,G,T}, the objective is to model the probability distribution P(X)=∏t=1LP(xt∣x<t). To handle long genomic contexts efficiently, dnaHNet utilizes a recursive hierarchical architecture consisting of three primary differentiable modules: an Encoder (E), a Main Network (M), and a Decoder (D).
Refer to the framework diagram:
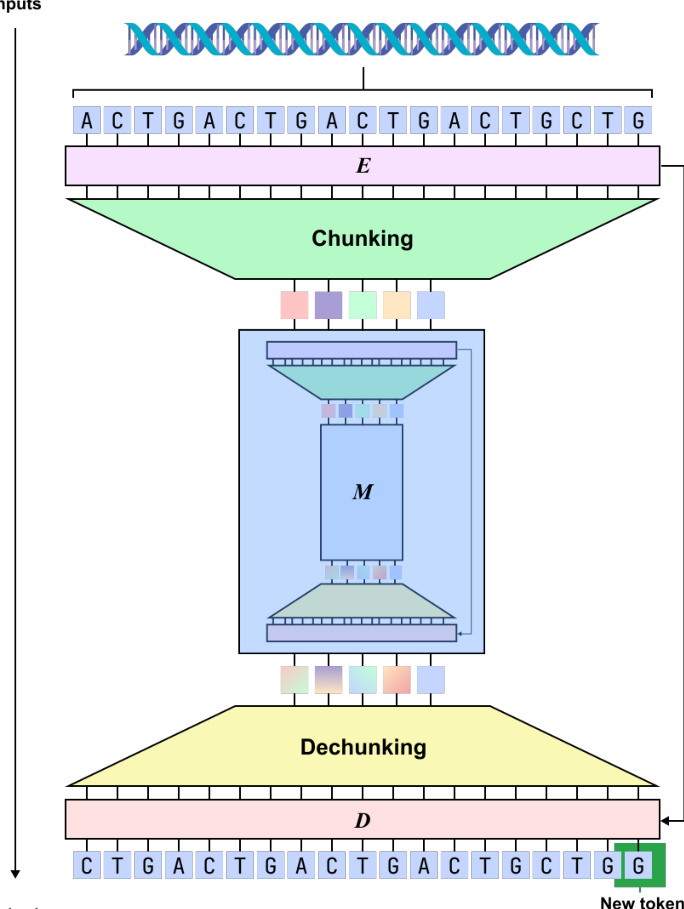
The Encoder is responsible for compressing nucleotide-level inputs into latent chunks through a dynamic segmentation mechanism. It employs a hybrid backbone consisting of four Mamba layers and one Transformer layer. The Encoder transforms input embeddings into hidden states h1:L∈RL×D. To determine segmentation boundaries, a boundary prediction module computes probabilities pt∈[0,1] using the following formulation: \npt=21(1−CosineSim(Wqht,Wkht−1)) where Wq and Wk are learnable projection matrices. High boundary probabilities are assigned to nucleotides with dissimilar representations, which encourages the model to segment at biologically significant transitions, such as codon boundaries. The Chunking layer then downsamples the output by selecting representations at these predicted boundaries, resulting in a compressed sequence E=(e1,…,eL′) where L′≤L.
As shown in the figure below:
The compressed sequence E is then processed by the Main Network M. This module can be a standard Transformer or another H-Net module, which allows for recursive chunking to capture multiple levels of abstraction. In a two-stage hierarchy, the first stage captures high-frequency local patterns like codon periodicity, while the second stage models long-range dependencies across functional regions. The Main Network outputs processed latent states E^=(e^1,…,e^L′)∈RL′×D.
The Decoder D maps these latent states back to the original nucleotide resolution through a two-step process. First, a smoothing module refines the latent states into smoothed representations Eˉ=(eˉ1,…,eˉL′) using a recurrence that interpolates discrete chunks: eˉj=Pje^j+(1−Pj)eˉj−1 where Pj is the boundary probability for the j-th chunk. Second, an upsampler expands these smoothed latents to the original length L by copying the vector eˉc(t) to every nucleotide position t corresponding to the chunk index c(t). The Decoder then utilizes four Mamba layers and one Transformer layer to model autoregressive dependencies, with a linear head projecting the output to the nucleotide vocabulary logits to produce the next-nucleotide distribution.
The training process is conducted end-to-end using a composite objective. The primary component is the autoregressive next-token prediction loss: LNLL=−∑t=1LlogPθ(xt∣x<t) To prevent degenerate segmentation during training, the authors incorporate a ratio loss that regularizes the dynamic chunking toward a target downsampling ratio Rs for each stage s: Lrate(s)=Rs−1Rs((Rs−1)FsGs+(1−Fs)(1−Gs)) where Fs is the actual fraction of selected chunks and Gs is the average boundary probability. The total loss is defined as L=LNLL+α∑sLrate(s), where α is a regularization coefficient.
Experiment
The experiments compare dnaHNet against StripedHyena2 and Transformer++ architectures through scaling law analyses, zero-shot biological prediction tasks, and structural interpretability studies. Results demonstrate that dnaHNet achieves superior compute and inference efficiency, consistently outperforming baselines in perplexity scaling and downstream performance on protein variant effect and gene essentiality predictions. Furthermore, the model demonstrates an emergent ability to learn biological hierarchies without supervision, effectively discovering codon structures and functional genomic regions through its hierarchical compression mechanism.
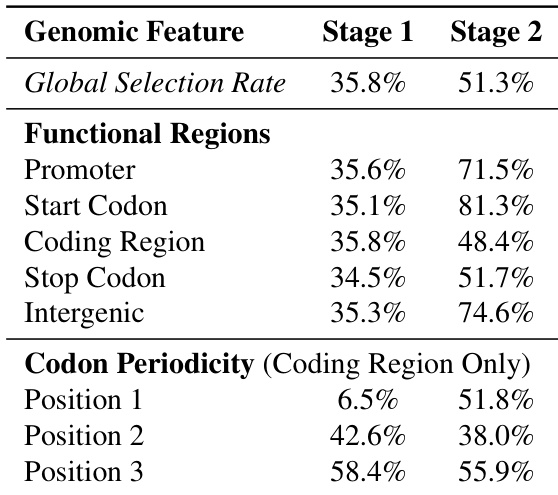
The authors analyze the learned hierarchical chunking boundaries of the dnaHNet model across different stages. The results demonstrate that the first stage captures local triplet codon structure, while the second stage identifies broader functional genomic organization. The first stage shows strong periodicity within coding regions, with selection rates varying significantly by codon position. The second stage exhibits higher selection rates for functional regions like promoters and intergenic areas compared to coding regions. The overall global selection rate increases from the first stage to the second stage.
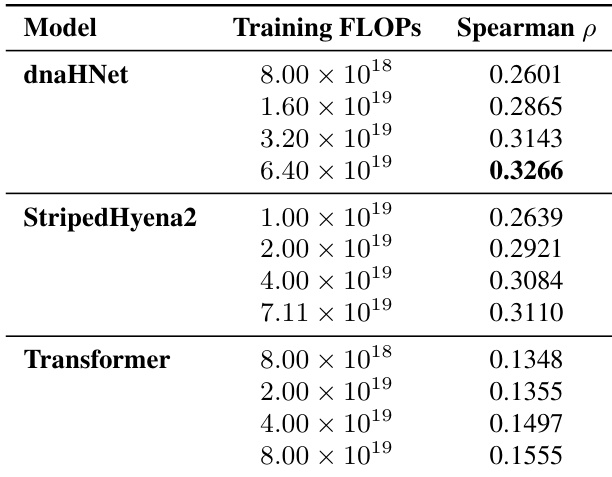
The authors evaluate the zero-shot protein variant effect prediction performance of dnaHNet against StripedHyena2 and Transformer baselines across various training compute budgets. Results show that dnaHNet consistently achieves higher Spearman correlation for predicting experimental fitness as training FLOPs increase. dnaHNet demonstrates superior predictive accuracy compared to both StripedHyena2 and Transformer architectures. The predictive performance of dnaHNet improves steadily as the amount of training compute increases. The performance gap between dnaHNet and the baseline models widens at higher training compute scales.
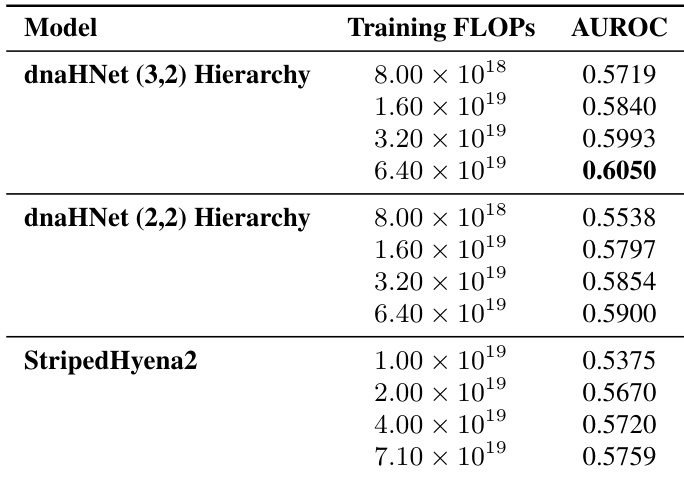
The authors compare the gene essentiality prediction performance of two dnaHNet hierarchical configurations against a StripedHyena2 baseline across various compute budgets. Results show that both dnaHNet architectures consistently outperform the baseline in AUROC as training compute increases. Both dnaHNet hierarchy versions demonstrate improved predictive accuracy as training FLOPs increase. The dnaHNet (3,2) hierarchy achieves higher AUROC scores compared to the (2,2) hierarchy at matched compute levels. dnaHNet models maintain a performance advantage over StripedHyena2 across the tested compute range.
The researchers evaluate the dnaHNet model by analyzing its hierarchical chunking boundaries and testing its predictive capabilities for protein variant effects and gene essentiality. The findings reveal that the model successfully captures both local codon structures and broader functional genomic organization through its multi-stage hierarchy. Across various compute budgets, dnaHNet consistently outperforms StripedHyena2 and Transformer baselines, demonstrating superior scaling and predictive accuracy in both protein fitness and gene essentiality tasks.