Command Palette
Search for a command to run...
VenusFactory: Eine einheitliche Plattform zur Datenabrufung für Proteinengineering und Feinabstimmung von Sprachmodellen
VenusFactory: Eine einheitliche Plattform zur Datenabrufung für Proteinengineering und Feinabstimmung von Sprachmodellen
Zusammenfassung
Natürliche Sprachverarbeitung (Natural Language Processing, NLP) hat wissenschaftliche Domänen jenseits der menschlichen Sprache erheblich beeinflusst, darunter auch die Proteinengineering, wo vortrainierte Protein-Sprachmodelle (Protein Language Models, PLMs) bemerkenswerte Erfolge erzielt haben. Dennoch bleibt die interdisziplinäre Anwendung aufgrund von Herausforderungen bei der Datensammlung, der Aufgabenbenchmarking und der praktischen Anwendung begrenzt. In dieser Arbeit präsentieren wir VenusFactory, eine vielseitige Engine, die die biologische Datenabfrage, standardisierte Aufgabenbenchmarking und modulare Fine-Tuning von PLMs integriert. VenusFactory unterstützt sowohl die Informatik- als auch die Biologie-Community durch die Auswahl zwischen einer Befehlszeilen-Execution und einer no-code-Schnittstelle basierend auf Gradio. Die Plattform integriert über 40 proteinrelevante Datensätze und über 40 gängige PLMs. Alle Implementierungen sind auf https://github.com/tyang816/VenusFactory opensourced.
One-sentence Summary
The authors from Shanghai Jiao Tong University, Shanghai Artificial Intelligence Laboratory, and East China University of Science and Technology present VENUSFACTORY, a unified platform integrating biological data retrieval, standardized benchmarking, and modular fine-tuning of 40+ protein language models, enabling both code-based and no-code access for interdisciplinary protein engineering applications.
Key Contributions
-
Addressing the gap in interdisciplinary collaboration, VENUSFACTORY integrates biological data retrieval from major databases like RCSB PDB, UniProt, and AlphaFold DB, enabling efficient, bulk downloading and standardized formatting for both computational and biological researchers.
-
The platform introduces a unified framework for standardized task benchmarking and modular fine-tuning of over 40 pre-trained protein language models (PLMs), supporting diverse methods including LoRA, Freeze & Full fine-tuning, and SES-Adapter across 40+ protein-related downstream tasks.
-
VENUSFACTORY provides accessible interfaces—both a no-code Gradio web UI and a command-line tool—enabling seamless model training, evaluation, and application, with all components open-sourced on GitHub and Hugging Face under the Apache 2.0 license.
Introduction
The authors leverage the growing success of protein language models (PLMs) in enzyme engineering to address persistent barriers in interdisciplinary collaboration between computer science and biology. Despite advances in PLMs like ESM2 and their application in tasks such as function prediction and sequence design, prior work suffers from fragmented data access, inconsistent benchmarking, and steep technical barriers that limit usability for non-experts. Existing tools either focus narrowly on data integration or lack comprehensive support for fine-tuning and evaluation across diverse protein tasks. To overcome these challenges, the authors introduce VENUSFACTORY, a unified platform that integrates high-throughput data retrieval from major biological databases, standardized benchmarking across 40+ datasets, and modular fine-tuning of 40+ PLMs via both a no-code Gradio interface and command-line tools. By providing a reproducible, open-source framework built on PyTorch and hosted on Hugging Face, VENUSFACTORY lowers the entry barrier for researchers across disciplines, enabling seamless adoption of state-of-the-art AI methods in protein engineering.
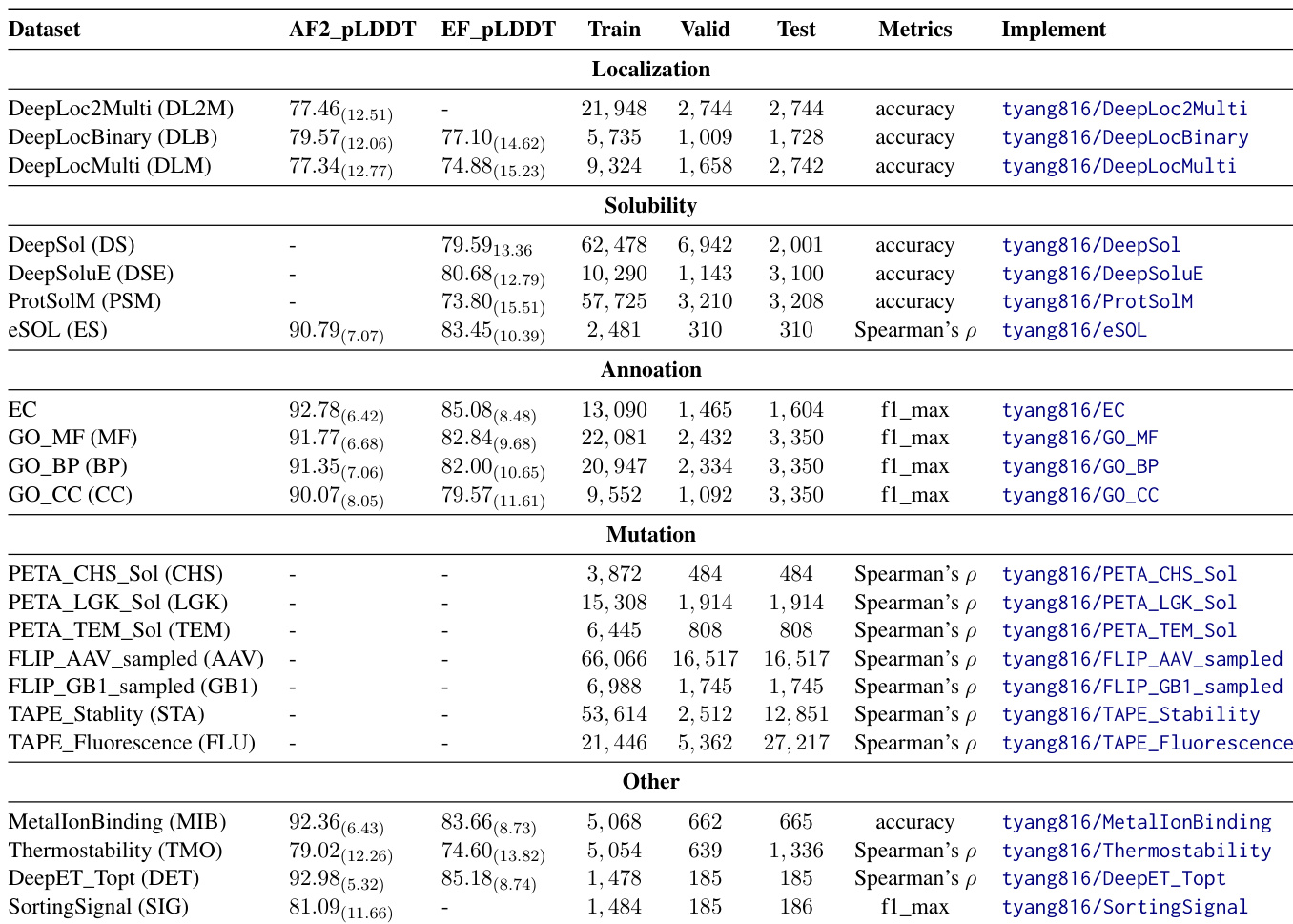
Dataset
-
The dataset is composed of data from four major protein databanks: RCSB PDB (over 200,000 experimentally determined 3D structures), UniProt (over 250 million protein sequences and functional annotations), InterPro (approximately 41,000 proteins with family, domain, and functional site annotations), and AlphaFold DB (AlphaFold2-predicted structures linked to UniProt IDs).
-
Each benchmark subset is curated for specific bioengineering tasks:
- Localization: Includes DeepLocBinary (binary membrane association), DeepLocMulti (multi-class localization), and DeepLoc2Multi (multi-label, multi-class), all with sequence and AlphaFold2/ESMFold-predicted structures.
- Solubility: Features three binary classification datasets (DeepSol, DeepSoluE, ProtSolM) and one regression benchmark (eSol), with ESMFold-predicted structures; eSol also includes AlphaFold2-predicted structures.
- Annotation: Four multi-class, multi-label benchmarks (EC, GO-CC, GO-BP, GO-MF) using Enzyme Commission and Gene Ontology annotations, with structures from AlphaFold2 and ESMFold.
- Mutation: 19 datasets with numeric labels for regression, including enzyme solubility (PETA_TEM_Sol, PETA_CHS_Sol, PETA_LGK_Sol), fluorescence/stability (TAPE_Fluorescence, TAPE_Stability), and viral fitness (FLIP_AAV) and nucleotide-binding (FLIP_GB1) benchmarks, each with defined splitting rules.
- Other Properties: Five additional datasets for thermostability, optimal enzyme temperature (DeepET_Topt), metal ion binding, and sorting signal detection, all using AlphaFold2-predicted structures, with ESMFold used in thermostability, DeepET_Topt, and sorting signal tasks.
-
The data is processed through multithreaded downloading using simulated HTTP requests with user-agent spoofing and concurrent execution, enabling efficient retrieval from UniProt, AlphaFold DB, and RCSB PDB in formats like .cif, .pdb, .xml, and .json. Metadata is indexed by RCSB ID or UniProt ID and stored in structured JSON format.
-
Protein structures are serialized into discrete tokens using three methods: DSSP (3- or 8-class secondary structure), FOLDSEEK (20D 3Di tokens via VQ-VAE), and ESM3 (4,096D integer representations from local subgraphs).
-
The dataset is used for training and evaluation across over 40 pre-trained protein language models (PLMs), including ESM2, ESM-1b, ProtBERT, ANKH, and ProtT5. Training employs both standard sequence truncation and a non-truncating approach that dynamically determines optimal token limits per batch to preserve sequence integrity.
-
Data is standardized across all benchmarks with consistent formats and includes evaluation metrics such as accuracy, F1-score, and Spearman’s correlation. Confidence scores (pLDDT) from AlphaFold2 and ESMFold are reported with mean and standard deviation.
-
Metadata, sequences, and structures are stored hierarchically for easy access, with InterPro data split into domain details, accession metadata, and UniProt ID lists, while UniProt sequences are saved in FASTA and AlphaFold structures are organized by ID prefix.
-
The system includes robust error handling, logging failed downloads in "failed.txt" for retries, and uses caching and adaptive rate limiting to prevent API overuse and improve retrieval efficiency.
Method
The authors leverage a modular architecture in VenusFactory to support diverse biological system analysis tasks, structured around three primary components: Collection, Benchmarking, and Application. The framework begins with the Collection module, which interfaces with external biological databases such as PDB, UniProt, InterPro, and AlphaFold. This module employs multithreaded downloading for efficient data retrieval and includes a processing module for handling data serialization and format conversion. As shown in the figure below, the collected data is then directed to the Benchmarking module, which organizes it into structured formats for downstream tasks. This includes data acquisition for prediction tasks such as localization, solubility, mutation effect, function annotation, and other properties, with inputs categorized by sequence, structure, attributes, and labels.
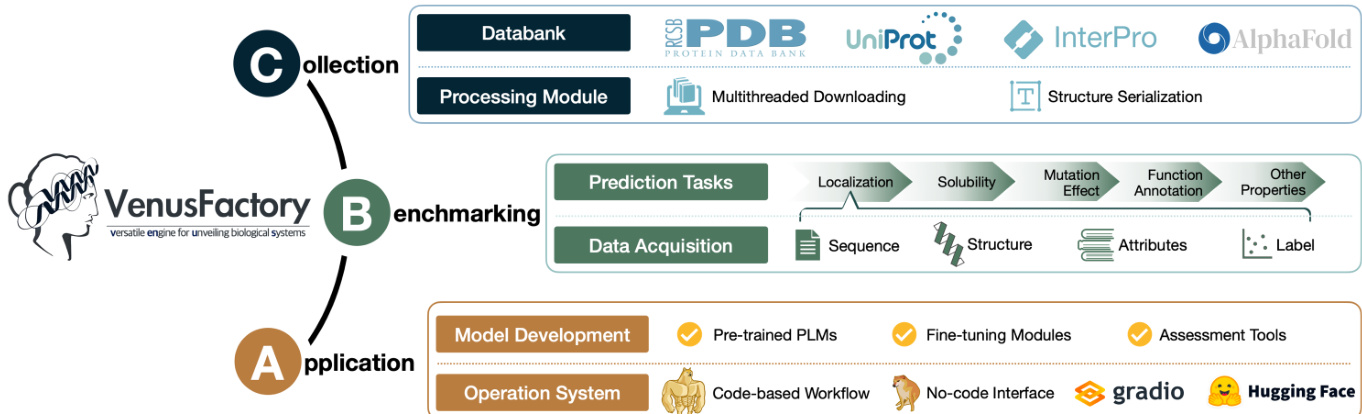
The Application layer integrates Model Development and Operation System components. Model Development encompasses pre-trained protein language models (PLMs), fine-tuning modules, and assessment tools. For fine-tuning, VenusFactory supports both classic approaches—freeze fine-tuning, which updates only the readout layers while keeping PLM parameters fixed, and full fine-tuning, which updates the entire model. Additionally, it incorporates parameter-efficient methods such as LoRA and its variants, as well as the protein-specific SES-ADAPTER, which utilizes cross-attention between PLM representations and sequence-structure embeddings (e.g., from FOLDSEEK) to enhance task-specific performance. The framework also supports multiple classification heads, including a two-layer fully connected network with average pooling, dropout, and GeLU activation; a lightweight head combining 1D convolutional feature extraction with attention-weighted pooling; and ATTENTION1D, which employs masked 1D convolution-based attention pooling and a non-linear projection layer for multi-class classification. The Operation System provides a code-based workflow and a no-code interface, with integration to platforms such as Gradio and Hugging Face for streamlined deployment and accessibility.
Experiment
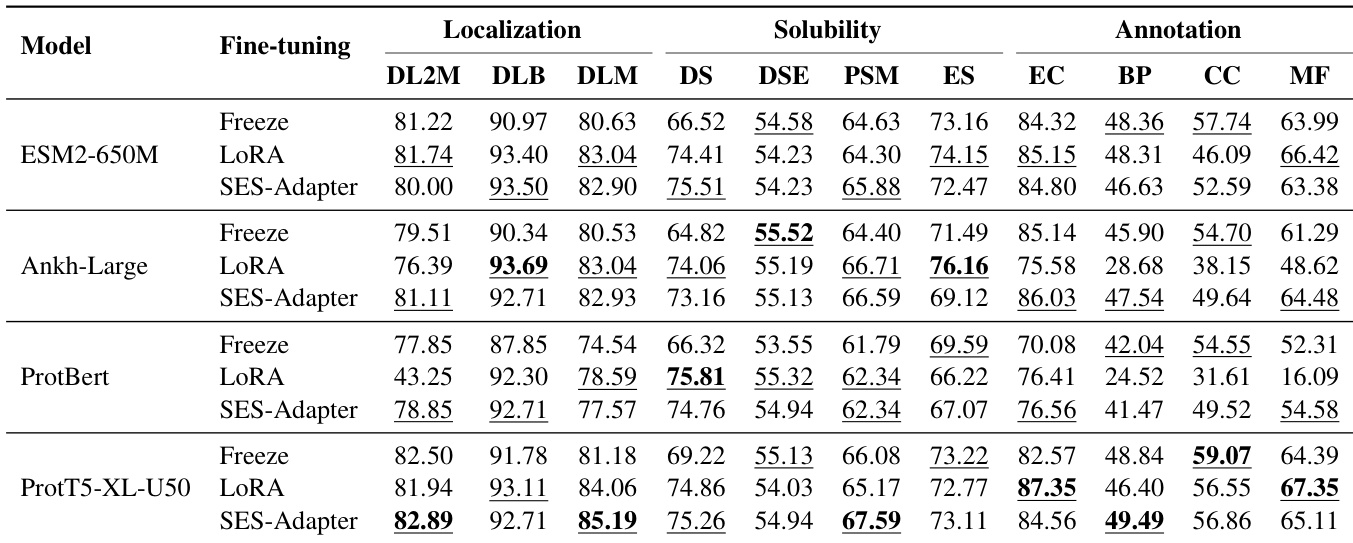
- Evaluated multiple pre-trained language models (PLMs) with three fine-tuning strategies—Freeze, LoRA, and SES-ADAPTER—across diverse downstream tasks including solubility prediction, mutation effect analysis, localization, and functional annotation.
- SES-ADAPTER consistently outperformed other methods, achieving the highest results in solubility prediction (DSE, PSM) and mutation effect prediction (AAV, GB1), demonstrating superior robustness across tasks.
- PROT5-XL-U50 achieved the best overall performance, excelling in annotation and mutation prediction, while PROTBERT showed limitations in mutation and certain annotation tasks.
- LoRA performed strongly in localization tasks (e.g., highest score on DLB) but lacked consistency across solubility and annotation benchmarks.
- Freeze method yielded the lowest performance, especially in annotation tasks (BP, MF), highlighting the necessity of full or lightweight fine-tuning for optimal model performance.
- On multiple datasets, SES-ADAPTER achieved state-of-the-art results, with significant improvements in key metrics such as F1-max for multi-label classification and Spearman’s ρ for ranking tasks.
- Training used ADAMW optimizer with a learning rate of 0.0005, 12,000-token batch limit, gradient accumulation of 8, and early stopping after 10 epochs of no improvement, ensuring reproducibility with seed 3407.
- Experiments conducted on 20 RTX 3090 GPUs over two months, with structural inputs derived from FOLDSEEK and DSSP 8-class representations for SES-ADAPTER.
The authors use a standardized experimental setup to evaluate various protein language models across multiple tasks, with fine-tuning methods including Freeze, LoRA, and SES-ADAPTER. Results show that SES-ADAPTER consistently outperforms other methods, particularly in solubility and mutation prediction, while model choice and task-specific fine-tuning strategies significantly impact performance.
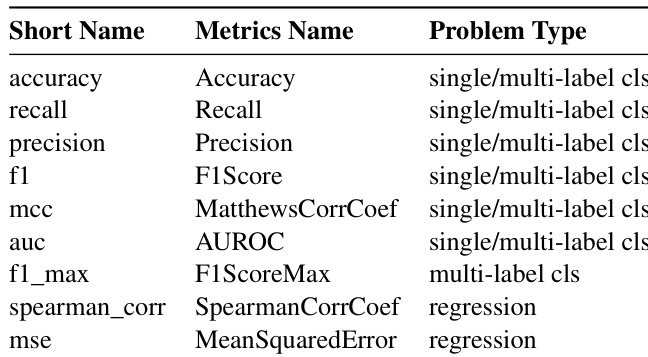
The authors use a range of evaluation metrics to assess model performance across different problem types, including classification and regression tasks. Results show that for classification, metrics such as accuracy, precision, recall, F1-score, and AUROC are used, while for regression tasks, Spearman's correlation and mean squared error are applied.
Results show that SES-Adapter consistently outperforms Freeze and LoRA across all models and tasks, particularly in mutation prediction and other downstream applications. Among the models, ProtT5-XL-U50 achieves the highest overall performance, while SES-Adapter demonstrates robustness and effectiveness across different fine-tuning strategies.
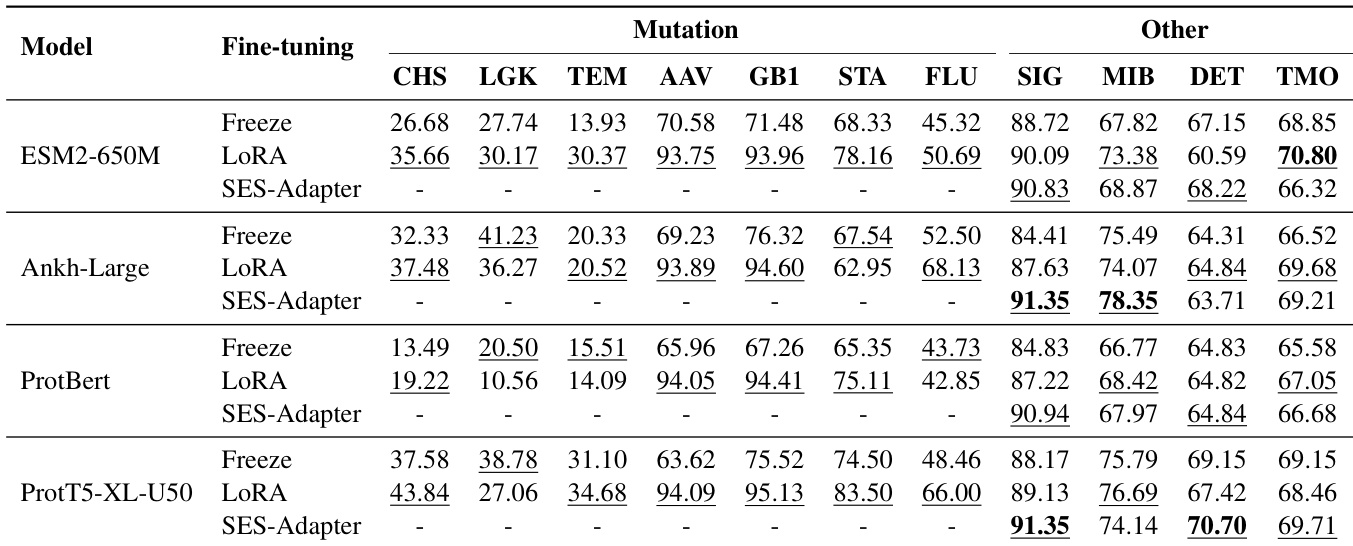
The authors evaluate multiple protein language models using different fine-tuning strategies across various downstream tasks, with results showing that SES-ADAPTER consistently outperforms other methods, particularly in solubility and mutation prediction. The table highlights that PROT5-XL-U50 achieves the highest overall performance, while the Freeze method yields the lowest results, especially in annotation tasks, indicating the necessity of adaptive fine-tuning for optimal model performance.
The authors use three fine-tuning methods—Freeze, LoRA, and SES-Adapter—to evaluate four protein language models across localization, solubility, and annotation tasks. Results show that SES-Adapter consistently outperforms the other methods, particularly in solubility and mutation prediction, while LoRA achieves the highest scores in localization tasks, and Freeze generally yields the lowest performance across most benchmarks.