Command Palette
Search for a command to run...
Multimodale Modelle Beschleunigen Die Zuordnung Neuer Materialien Zu Industriellen Anwendungen Und Sagen Materialeigenschaften Voraus, Ohne Dass Eine Vollständige Kristallstruktur Erforderlich ist.
Südlich des Huai-Flusses wachsen Orangen zu Orangen, nördlich davon zu Mandarinen. Derselbe Samen bringt in unterschiedlichen Böden und Klimazonen völlig unterschiedliche Früchte hervor. Dieses natürliche Wachstumsgesetz zeigt sich auch in der Materialchemie: Neue Materialien zeigen für unterschiedliche Anwendungen unterschiedliche Eigenschaften. Forschungsergebnisse belegen, dass Wissenschaftler jedes Jahr Hunderttausende neuer Materialien entwickeln. Diese sind wie unzählige „Samen“ mit immensem Potenzial, die in einer geeigneten Umgebung Wurzeln schlagen müssen, um zu gedeihen.
Obwohl neue Materialdesigns häufig für spezifische Anwendungen synthetisiert werden, bieten sie oft auch in anderen Bereichen Einsatzmöglichkeiten. Die schnelle Bestimmung der Anwendungsszenarien neuer Materialien bleibt jedoch eine anspruchsvolle Aufgabe.Ein Beispiel hierfür sind die weit verbreiteten kristallinen Materialien, die sogenannten Metall-organischen Gerüstverbindungen (MOFs). Ihre wichtigste Anwendung ist die Speicherung von Gasen wie Wasserstoff und Methan. Sie zeigen zudem ein hervorragendes Leistungspotenzial in Membranen, Dünnschichtgeräten, der Katalyse und der biomedizinischen Bildgebung. Traditionelle Ansätze zur Bestimmung der optimalen Anwendung von MOFs basieren auf Materialeigenschaften als Zwischenbasis, deren Prüfung jedoch kostspielig ist (hinsichtlich Zeit, Ausrüstung und Fachwissen). Darüber hinaus benötigen computergestützte Screening- und maschinelle Lernmethoden die vollständige Kristallstruktur zur Vorhersage von Eigenschaften. Die Kristallstrukturanalyse ist jedoch zeitaufwändig und nach der MOF-Synthese nicht ohne Weiteres verfügbar.
Um dieses Problem zu lösen, hat ein Forschungsteam der Abteilung für Chemieingenieurwesen und angewandte Chemie der Universität Toronto, Kanada, eine neue Methode vorgeschlagen, die auf einem multimodalen Modell des maschinellen Lernens basiert.Nutzung der nach der MOF-Synthese verfügbaren Informationen zur Vorhersage ihrer potenziellen Eigenschaften und Verwendungsmöglichkeiten,Das Modell umfasst beispielsweise Pulver-Röntgenbeugungsmuster (PXRD) und die bei der Synthese verwendeten Chemikalien. Das Forschungsteam ergänzte das Modell um ein Anwendungsempfehlungssystem, das unmittelbar nach der MOF-Synthese Anwendungsvorschläge liefert. Diese Forschung beschleunigt die Verbindung zwischen der Synthese von Metall-organischen Gerüstverbindungen (MOFs) und ihren Anwendungsszenarien.
Die entsprechende Forschungsarbeit wurde in Nature Communications unter dem Titel „Connecting metal-organic framework synthesis to applications using multimodal machine learning“ veröffentlicht.
Forschungshighlights:
* Diese Methode nutzt ausschließlich nach der Synthese verfügbare Informationen, um die potenziellen Eigenschaften und Verwendungsmöglichkeiten von MOFs vorherzusagen. Sie gibt Anwendungsempfehlungen unmittelbar nach der Synthese der MOFs ab und verkürzt so den Zyklus von der Materialsynthese bis zur Anwendung erheblich.
* Die Vorhersageleistung des Modells ist mit der von fortgeschrittenen Modellen vergleichbar, die präzise Kristallstrukturdaten erfordern (z. B. CGCNN und MOFormer), und übertrifft diese unter bestimmten Bedingungen sogar. Es ist auch angesichts von experimentellem Rauschen, Kristallstrukturdefekten und anderen Bedingungen stabil und zuverlässig und weist eine hervorragende Robustheit auf.
* Diese Studie kombiniert ein visuelles Anwendungsempfehlungssystem, um ein Synthese-Vorhersage-Anwendungs-Closed-Loop-System aufzubauen;
Papieradresse:
https://www.nature.com/articles/s41467-025-60796-0
Folgen Sie dem offiziellen Konto und antworten Sie mit „MOFs“, um das vollständige PDF zu erhalten
Weitere Artikel zu den Grenzen der KI: https://hyper.ai/papers
„Daten sind der Syntheseort“: MOFs-Datenkonstruktionsstrategie zur Anwendungsvorhersage
In dieser Studie wurden insgesamt 6 Datenbanken mit metallorganischen Gerüsten (MOFs) für das Training und die Bewertung des Modells verwendet: CoRE-2019, BW20K, ARABG, QMOF, hMOF und CSD-Teilmengen.In:
* hMOF bietet eine extrem große Bibliothek hypothetischer Strukturen, die zur Verbesserung der Generalisierungsfähigkeit des Modells beiträgt.
* BW20K und ARABG werden verwendet, um die Vielfalt zu erhöhen und Aufgaben mit wenigen Schüssen zu unterstützen.
* Die CSD-Teilmenge wird verwendet, um die Robustheit des Modells unter experimenteller Verzerrung zu testen.
Das Forschungsteam verwendete Kristallstrukturen aus den Datenbanken CoRE 2019, BW20K, ARABG, QMOF und hMOF und berechnete mithilfe des Pymatgen-XRD-Moduls simulierte PXRD-Muster von 0 bis 90 Grad, um die nach der Synthese in tatsächlichen Experimenten erhaltenen strukturellen Charakterisierungsinformationen zu simulieren. Informationen zu chemischen Vorläufern, bestehend aus Metallknoten und organischen Linkern, wurden im Format [Metalltyp].[organischer Linker] erstellt und zur Wortsegmentierung in den Transformer-Kanal des Modells eingegeben.
Ein multimodales Lernframework, das durch selbstüberwachtes Vortraining gesteuert wird
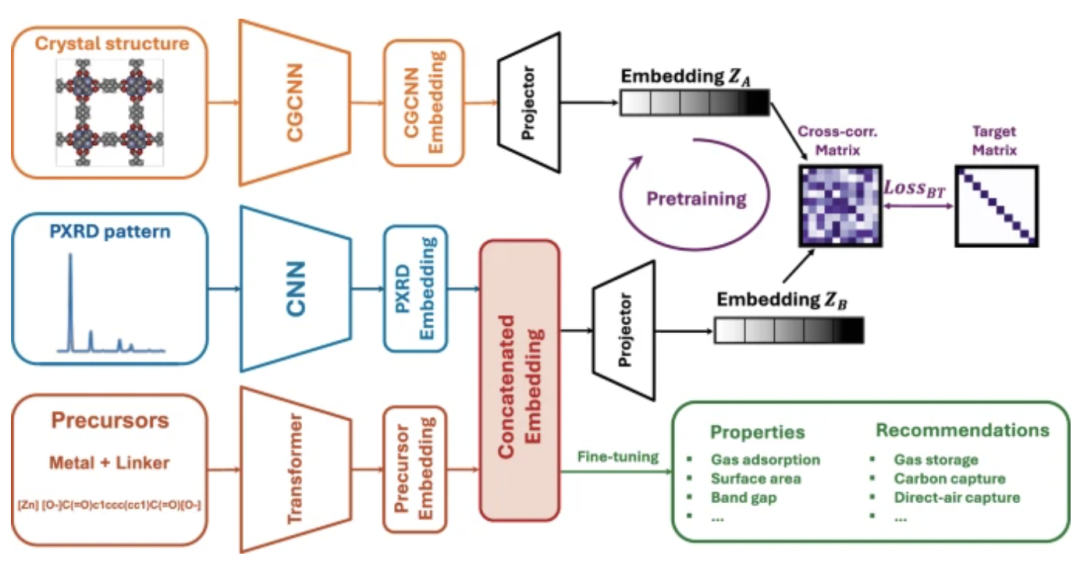
Das Forschungsteam schlug ein selbstüberwachtes, vorab trainiertes, multimodales Lernframework vor, mit dem Ziel, die Abhängigkeit von der vollständigen Kristallstruktur zu beseitigen.Sagen Sie die Eigenschaften und potenziellen Anwendungen von MOFs voraus, indem Sie nur die nach der Synthese verfügbaren Informationen verwenden.
Der Arbeitsablauf dieses selbstüberwachten multimodalen Modells ist in der folgenden Abbildung dargestellt. Ein Präkursor-String und ein Pulver-Röntgenbeugungsspektrum (PXRD) dienen als Eingaben, werden über einen Transformer bzw. ein Convolutional Neural Network (CNN) eingebettet und zur Feinabstimmung an einen Regressionskopf übergeben. Der Präkursor liefert Informationen über die chemischen Eigenschaften des Materials, während das PXRD-Muster zusätzliche Informationen über die geometrische Gesamtstruktur liefert.
Die von Transformer kodierten chemischen Vorläuferstrings und die von CNN verarbeiteten PXRD-Spektren werden durch Merkmalsspleißung und -projektion in einen einheitlichen Darstellungsraum konstruiert.Um den Mangel von „Vorläufer + PXRD“ auszugleichen, der darin besteht, die lokale chemische Umgebung nicht direkt charakterisieren zu können, führte das Forschungsteam einen selbstüberwachten Vortrainingsmechanismus ein.Die Modellausgabe wird mit der Einbettung des Crystal Graph Convolutional Neural Network (CGCNN) ausgerichtet, und die Kreuzkorrelationsmatrix wird durch den Barlow-Zwillings-Verlust so eingeschränkt, dass sie nahe an der Identitätsmatrix liegt, wodurch das Modell dazu angeleitet wird, die Ausdruckskraft der lokalen chemischen Umgebung zu erlernen.
Auf dieser Grundlage kann das Modell nach selbstüberwachtem Training mit umfangreichen, unmarkierten Daten schnell mit begrenzten markierten Proben konvergieren und hochpräzise Vorhersagen der Porenstruktur, der chemischen Abhängigkeitseigenschaften und der quantenchemischen Eigenschaften erzielen.
Insbesondere basierend auf den Kristallstrukturen aus der MOF-Datenbank,Auch bei kleinen Datenmengen können mit dieser Methode verschiedene Eigenschaften präzise vorhergesagt werden.Dazu gehören die Porenstruktur, chemisch abhängige Eigenschaften und quantenchemische Eigenschaften.
Während der Selbstüberwachungs- und Trainingsphase wurde eine SSL-Pipeline (Self-Supervised Learning) erstellt, um Repräsentationslernen zwischen dem Crystal Graph Convolutional Neural Network (CGCNN) und dem Modell durchzuführen. Dadurch wurde die Einschränkung des Modells überwunden, die lokale Umgebung des MOF nicht anhand seiner Eingaben verstehen zu können. Die Gewichte des Modells wurden initialisiert, was eine schnelle Konvergenz zu einer Lösung ermöglichte. Das selbstüberwachte Lernen wurde an den CGCNN-Einbettungen durchgeführt. Jede Einbettung der Größe 512 wurde aus dem CGCNN und dem Projektor des Modells extrahiert und eine Kreuzkorrelationsmatrix der Form (512, 512) erstellt. Die Barlow-Twin-Verlustfunktion wurde verwendet, um die Differenz zu minimieren und die Kreuzkorrelationsmatrix nahe an die Identitätsmatrix zu bringen, wodurch Repräsentationslernen erreicht wurde.
Evaluation multimodaler Modelle
Um zu beweisen, dass das Modell verschiedene MOF-Eigenschaften effektiv vorhersagen und die Grundlage für die Kombination von MOF-Synthese und -Anwendung legen kann, bewertete das Forschungsteam die Genauigkeit des Modells mithilfe des Spearman-Rangkorrelationskoeffizienten (SRCC) und des mittleren absoluten Fehlers (MAE), um die Vorhersagegenauigkeit des Modells bei geometrisch abhängigen Eigenschaften, chemisch abhängigen Eigenschaften und quantenchemischen Eigenschaften zu bewerten, und führte Benchmark-Vergleiche mit CGCNN, MOFormer und deskriptorbasierten Modellen des maschinellen Lernens durch.
Die Ergebnisse zeigen, dassDie Modellgenauigkeit dieses Modells ist vergleichbar mit der von Modellen, die auf der vollständigen Kristallstruktur beruhen.Es übertrifft sogar CGCNN und MOFormer in der geometrischen Leistung und bestätigt damit, dass eine hochpräzise Eigenschaftsvorhersage nur unter Verwendung von Syntheseinformationen erreicht werden kann. Damit wird eine experimentelle Grundlage für die schnelle Anpassung der MOF-Synthese an die Anwendung geschaffen.
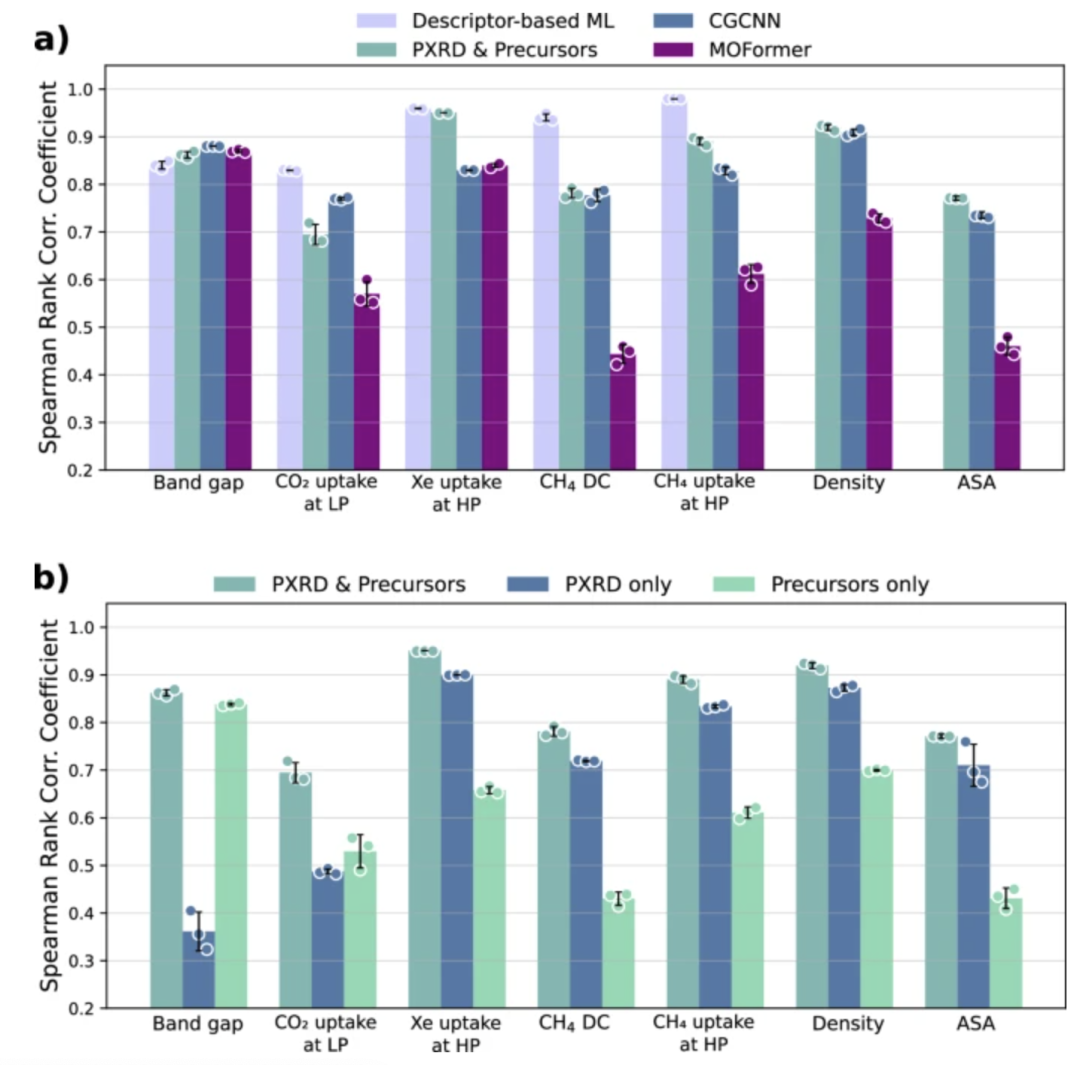
Darüber hinaus führte das Team Ablationsexperimente durch und verglich Modelle, die ausschließlich auf chemischen Vorläufern und Modelle, die ausschließlich auf PXRD beruhten, mit dem in dieser Studie entwickelten multimodalen Modell. Die Ergebnisse zeigten, dass das Modell, das nur chemische Vorläufer als Eingabe akzeptierte, die gesamte MOF-Struktur nicht effektiv erfassen konnte und bei geometriebezogenen und rein geometrischen Eigenschaften schlecht abschnitt. Während das Modell, das nur PXRD akzeptierte, die gesamte MOF-Struktur gut erfasste, konnte es die lokale Umgebung nicht widerspiegeln, was zu schlechten Ergebnissen bei chemisch bezogenen und quantenchemischen Eigenschaften (wie CO₂-Adsorption bei niedrigem Druck und Bandlücke) führte. Beide Modelle wiesen Einschränkungen auf. Die Ergebnisse zeigen, dass das multimodale Modell nur durch die Kombination von PXRD (das geometrische Informationen liefert) mit der Vorläuferkette (die chemische Informationen liefert) umfassende und genaue Vorhersagen für alle drei Eigenschaftskategorien erreichen kann; die Verwendung einer der beiden Modalitäten allein schneidet eindeutig schlechter ab.
Überprüfung der Modellstabilität: Bewertung der Robustheit gegenüber strukturellen Fehlern und experimentellem Rauschen
Stabilität ist ein wichtiges Maß für die Bewertung der Zuverlässigkeit eines maschinellen Lernmodells in realen Szenarien. Zu diesem Zweck bewertete das Forschungsteam systematisch die Robustheit des vorgeschlagenen multimodalen Modells unter nicht idealen Bedingungen. Zunächst verwendeten die Forscher experimentelle Kristallstrukturen aus der Cambridge Structural Database (CSD), um die entsprechenden PXRD-Muster zu berechnen und so gängige Strukturabweichungen in realen Experimenten zu simulieren, wie beispielsweise fehlende Wasserstoffatome und das Vorhandensein gebundener oder ungebundener Lösungsmittel. Die Bewertung konzentrierte sich auf eine einzige geometriebezogene Eigenschaft: die Vorhersage der Methanadsorptionskapazität unter hohem Druck für Methanspeicheranwendungen.
Die Ergebnisse zeigen, dassDas Modell kann unter den oben genannten Variationsbedingungen immer noch eine gute Vorhersagefähigkeit aufrechterhalten.Die Rangfolge der CH₄-Hochdruckadsorptionsleistung weist eine starke Konsistenz auf und der relative Fehler wird unter 13% kontrolliert, was auf eine hohe Robustheit hinweist.
Auf dieser Grundlage führte das Team zusätzlich tatsächlich gemessene PXRD-Muster zu Tests ein, um die Stabilität des Modells angesichts tatsächlicher Messfehler wie Instrumentenrauschen und Temperaturschwankungen zu überprüfen. Obwohl es bei einigen Proben erhebliche Unterschiede zwischen den simulierten und experimentellen Mustern gibt, liefert das Modell in den meisten Fällen dennoch empfohlene Ergebnisse, die den simulierten Mustern nahe kommen und nur in Einzelfällen bei erheblichem Rauschen oder offensichtlicher Spitzenfehlausrichtung abweichen. In Kombination mit den oben genannten Experimenten zeigt sich, dass das multimodale Modell nicht nur unter idealen strukturellen Eingangsbedingungen eine hohe Vorhersagegenauigkeit aufweist, sondern auch bei unvollkommener experimenteller Struktur oder Rauschen im PXRD eine robuste Leistung beibehält.Seine breite Anwendbarkeit in der praktischen Materialforschung und -anwendung wurde nachgewiesen.
Die folgende Abbildung zeigt die Ergebnisse der Modellempfehlung und vergleicht die Unterschiede zwischen den simulierten PXRD-Mustern und den experimentellen PXRD-Mustern:
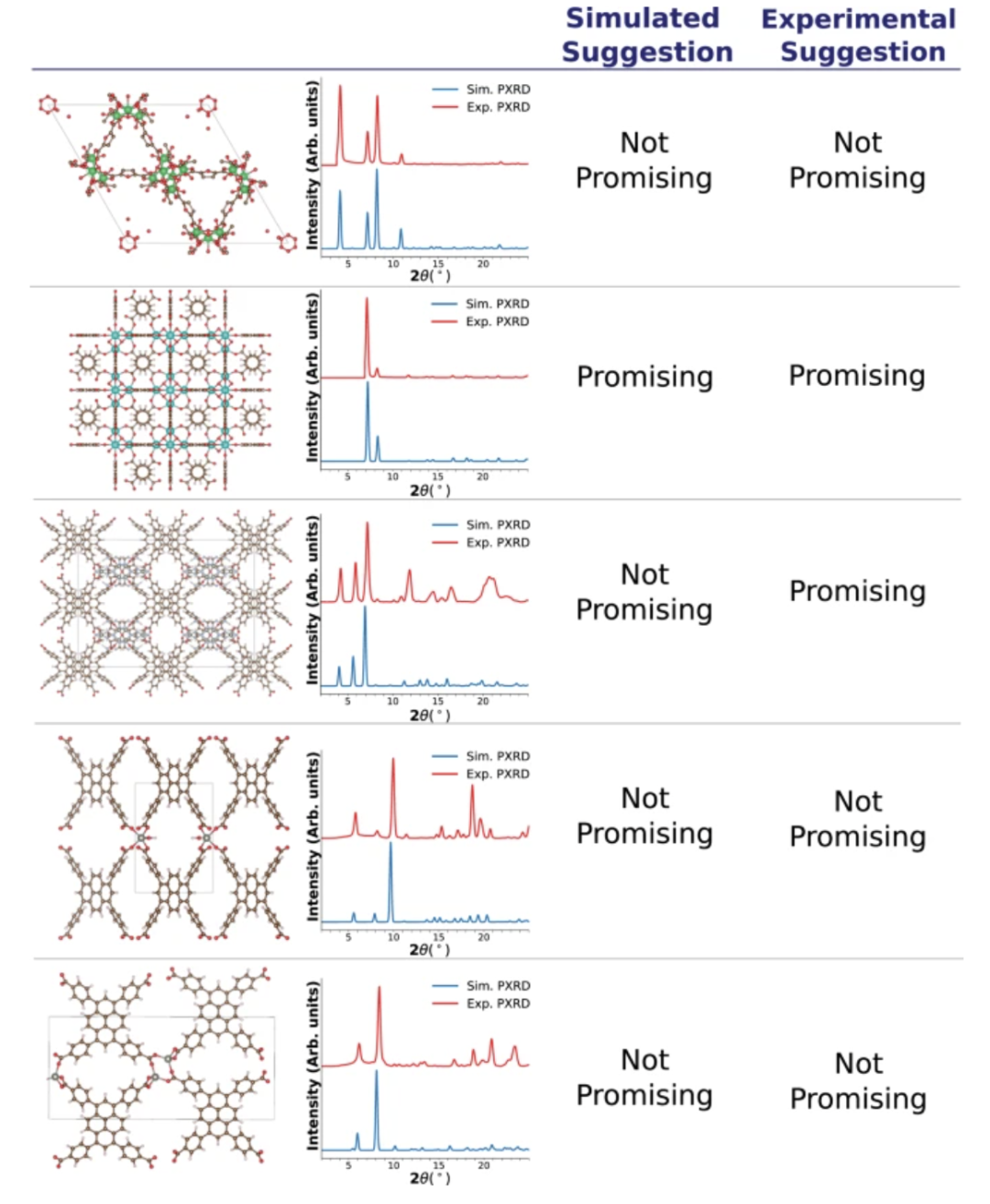
Synthese-Anwendung integriertes Empfehlungssystem
Basierend auf der beeindruckenden Leistung des Modells entwickelten die Forscher ein visuelles Empfehlungssystem für potenzielle Anwendungen, das neu synthetisierte MOFs anhand vorhergesagter Materialeigenschaften potenziellen Anwendungen (wie Gasspeicherung und Kohlenstoffabscheidung) zuordnet. Mithilfe der t-SNE-Technologie projiziert das System den latenten Raum des Modalmodells und stellt die empfohlenen Anwendungen für MOFs farblich dar. Die folgende Abbildung veranschaulicht die Zuordnung von Syntheseinformationen zu Anwendungsszenarien:
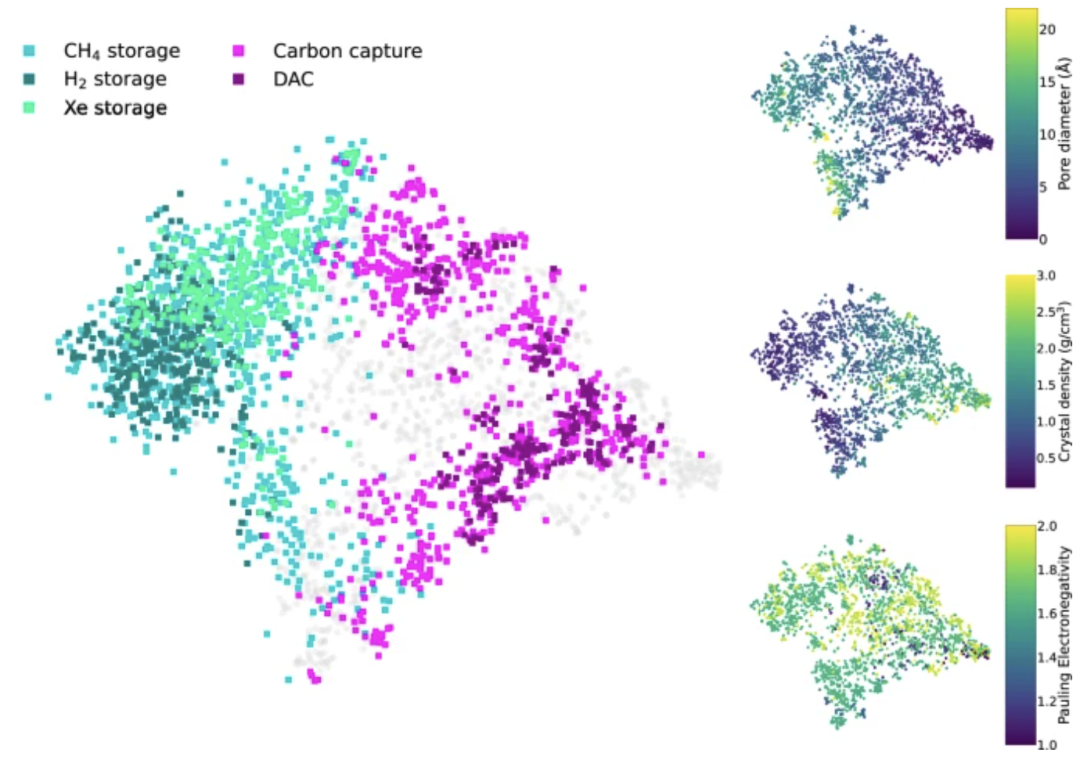
Um die Vorhersagefähigkeit des Modells für zukünftige Materialanwendungen zu überprüfen, führten die Forscher ein Zeitreiseexperiment durch.Das Modell wurde mit CoRE-2019-Einträgen trainiert, die vor 2017 in der CSD-Datenbank gespeichert waren, und mit Einträgen getestet, die nach 2017 gespeichert wurden, um Vorhersagen für zukünftige Materialien zu simulieren. Ziel des Experiments war es, die Leistung dieser MOFs in einer spezifischen Anwendung vorherzusagen: der Kohlendioxidadsorption. Die Ergebnisse zeigten, dass das Modell erfolgreich 18 MOFs mit Potenzial zur Kohlenstoffabscheidung identifizierte, von denen 15 ursprünglich für andere Anwendungen entwickelt wurden.
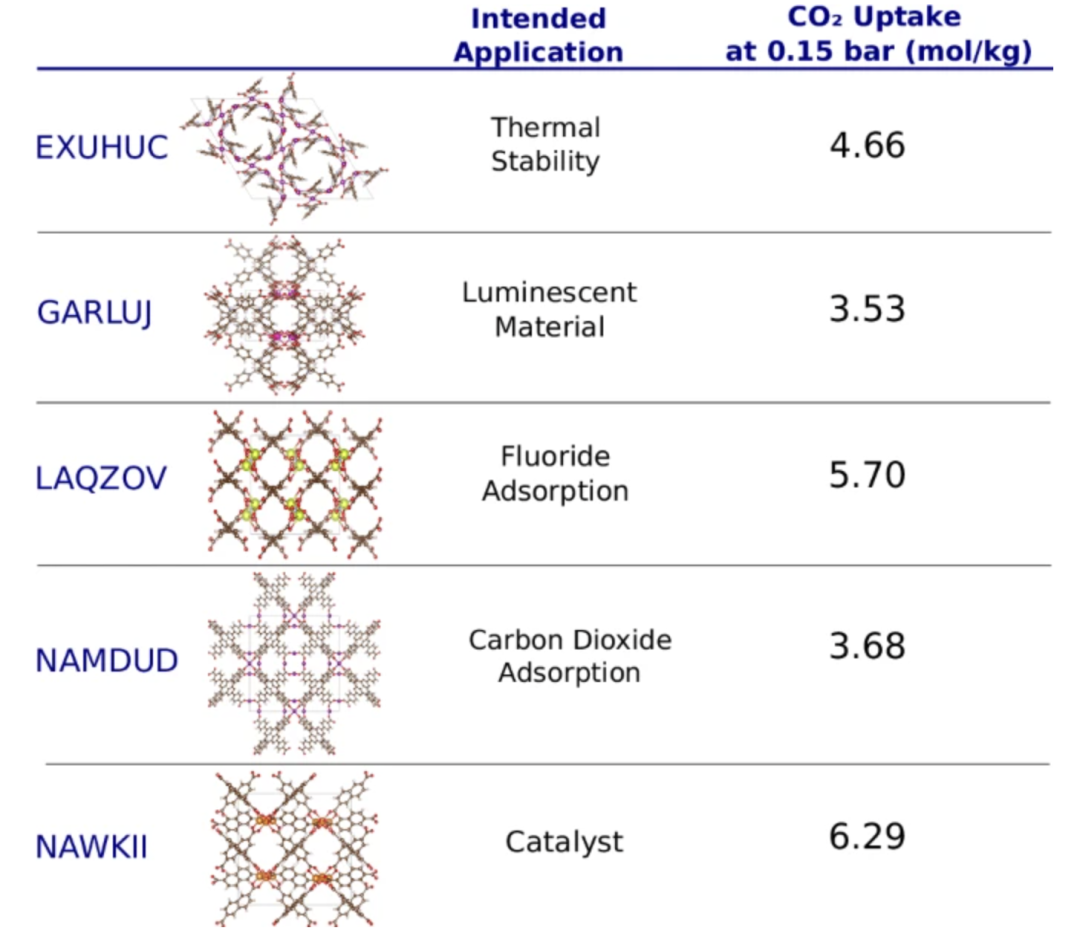
Einige dieser MOFs und ihre erwarteten synthetischen Anwendungen basieren auf den entsprechenden
Maschinelles Lernen revolutioniert die Materialwissenschaft
Dieser Artikel stellt einen multimodalen Ansatz des maschinellen Lernens vor, der die vielfältigen Eigenschaften von MOFs präzise vorhersagt und sie unabhängig von der Kristallstruktur spezifischen Anwendungen zuordnet. Dieser datengetriebene Trend breitet sich zeitlich und räumlich über ein immer breiteres Spektrum von Materialsystemen aus. So untersuchte beispielsweise das Team um Xie Jianxin und Su Yanjing von der University of Science and Technology Beijing die Anwendung interpretierbaren maschinellen Lernens in der Materialwissenschaft. Sie zeigten, dass die Kombination von Materialwissen mit maschinellem Lernen die Generalisierung und Vorhersagegenauigkeit des Modells deutlich verbessern kann und so neue Perspektiven für die Entwicklung der Materialwissenschaft eröffnet. Die zugehörige Forschung mit dem Titel „Interpretable Machine Learning Applications: A Promising Prospect of AI for Materials“ wurde in Advanced Functional Materials veröffentlicht.
Papieradresse:
https://advanced.onlinelibrary.wiley.com/doi/abs/10.1002/adfm.202507734
Ein Forschungsteam des Argonne National Laboratory in den USA hat ein generatives KI-Framework namens GHP-MOFsassemble vorgeschlagen, das neue MOF-Strukturen zufällig generieren und zusammensetzen kann. Dieses Framework nutzt molekulardynamische Simulationen, um nach hochstabilen MOFs zu suchen, und nutzt ein Crystal Graph Convolutional Neural Network (CGCNN) und Grand Canonical Monte Carlo-Simulationen, um die Kohlendioxid-Adsorptionskapazität der MOFs zu testen. Die zugehörige Forschung mit dem Titel „Ein generatives Framework für künstliche Intelligenz basierend auf einem molekularen Diffusionsmodell für die Entwicklung metallorganischer Gerüste zur Kohlenstoffabscheidung“ wurde in Communications Chemistry veröffentlicht.
Papieradresse:
https://www.nature.com/articles/s42004-023-01090-2
Ein Forschungsteam der Universität Oxford veröffentlichte eine Studie mit dem Titel „Der amorphe Zustand als Grenze im computergestützten Materialdesign“. Sie betont die entscheidende Rolle des maschinellen Lernens bei der Überwindung traditioneller Grenzen im Materialdesign. Die Studie zeigt, wie jüngste Fortschritte in der computergestützten Modellierung und künstlichen Intelligenz die bisher fehlende Verbindung zwischen der atomaren Struktur, den mikroskopischen Eigenschaften und der makroskopischen Funktionalität amorpher Festkörper schließen können.
Papieradresse:
https://www.nature.com/articles/s41578-024-00754-2
Diese Studienreihe zeichnet ein klares Bild: Die Materialwissenschaft tritt in ein neues Zeitalter der Intelligenz ein, und wir befinden uns mitten in einem durch maschinelles Lernen vorangetriebenen Wandel der Materialforschung. Noch wichtiger ist, dass sich die Intelligenz allmählich von der Entwicklung und Synthese neuer Materialien auf Anwendungsszenarien ausgeweitet hat, was den Einsatz neuer Materialien weiter vorantreiben wird.
Quellen:
1.https://pubs.acs.org/doi/10.1021/cr300014x
Erhalten Sie mit einem Klick hochwertige Papiere und ausführliche Interpretationsartikel im Bereich AI4S von 2023 bis 2024 ⬇️









