Command Palette
Search for a command to run...
Ausgewählt Für ICLR 2025! Das Von Der Universität Cambridge Vorgeschlagene Celcomen-Modell Ermöglicht Erstmals Die Identifizierbarkeit Kausaler Inferenz in Der Räumlichen Transkriptomik-Analyse
In der Biologie kodiert das Genexpressionsprofil einer Zelle Informationen sowohl über ihre intrinsischen Eigenschaften als auch über die äußere Gewebemikroumgebung. Um die komplexen Wechselwirkungen innerhalb und zwischen Zellen vollständig zu verstehen, ist es wichtig, die Ursache-Wirkungs-Beziehung zwischen diesen beiden Effekten zu entschlüsseln. Zu diesem Zweck ist ein robuster Rahmen zur kausalen Entflechtung erforderlich.
Kausale Entkopplung ist eine Methode des maschinellen Lernens, die darauf abzielt, nützliche Merkmale von irrelevanten Merkmalen zu trennen, indem kausale Beziehungen in Daten aufgedeckt werden. Dadurch wird die Abhängigkeit des Modells von Scheinkorrelationen verringert und die Robustheit und Generalisierungsfähigkeit des Modells verbessert. Neben der Entwicklung von Theorien des maschinellen Lernens, wie etwa der kausalen Entkopplung, haben technologische Fortschritte in der Biologie auch die Entwicklung der räumlichen Transkriptomik gefördert. Diese ermöglicht es Forschern, die Genexpression und die räumlichen Koordinaten von Zellen gleichzeitig mit Einzelzellauflösung zu messen und Störungsexperimente wie Gen-Knockout in großem Maßstab in räumlichen Proben durchzuführen.
Jedoch,Aktuelle rechnergestützte Ansätze zur räumlichen Transkriptomik vernachlässigen häufig die Modellierung kausaler Störungen auf Zell- und Gewebeebene.Dies ist von entscheidender Bedeutung, um die Mechanismen hinter Krankheitszuständen in Geweben aufzudecken. Beispielsweise kann das Modell „Virtual Cells“ die Auswirkungen von Änderungen in der Mikroumgebung und Makroumgebung (wie etwa Alter des Spenders, Zellgewebe, medikamentöse Behandlung, gRNA-vermittelter Gen-Knockout usw.) auf die Genexpression vorhersagen, und das Modell „Virtual Tissues“ kann nicht nur die Auswirkungen der Umgebung auf eine einzelne Zelle abschätzen, sondern auch auf die Auswirkungen einer einzelnen Zelle auf ihre umgebende Umgebung und das gesamte Gewebe schließen.
Auf dieser GrundlageEin Forschungsteam der Universität Cambridge hat ein virtuelles Gewebemodell namens Celcomen vorgeschlagen, bei dem es sich im Wesentlichen um ein neues, auf mathematischer Kausalität basierendes Graph-Neuralnetzwerk handelt, um die Geheimnisse der intrazellulären und interzellulären Genregulation in räumlichen Transkriptomik- und Einzelzelldaten zu entschlüsseln.Die Forscher bestätigten die Fähigkeit von Celcomen, Gen-Gen-Interaktionen sowohl in realen als auch in selbstsimulierten räumlichen Transkriptomikdaten zu entschlüsseln und wiederherzustellen.
Die entsprechenden Ergebnisse wurden für ICLR 2025 unter dem Titel „Estimation of single-cell and tissue perturbation effect in spatial transcriptomics via Spatial Causal Disentanglement“ ausgewählt.
Forschungshighlights:
* Die Studie belegt die Machbarkeit der Erweiterung des virtuellen Zellmodells auf das virtuelle Gewebemodell
* Die Studie schlägt das erste kausal identifizierbare Modell in der räumlichen Transkriptomik-Analyse vor
* Ableitung der Genregulation durch Integration dissoziierter Einzelzelldaten und räumlicher Einzelzelldaten
Papieradresse:
https://openreview.net/forum?id=Tqdsruwyac
Das Open-Source-Projekt „awesome-ai4s“ vereint mehr als 200 AI4S-Papierinterpretationen und stellt umfangreiche Datensätze und Tools bereit:
https://github.com/hyperai/awesome-ai4s
Datensatz: Erster Versuch, den Perturbmap-Datensatz zu verwenden
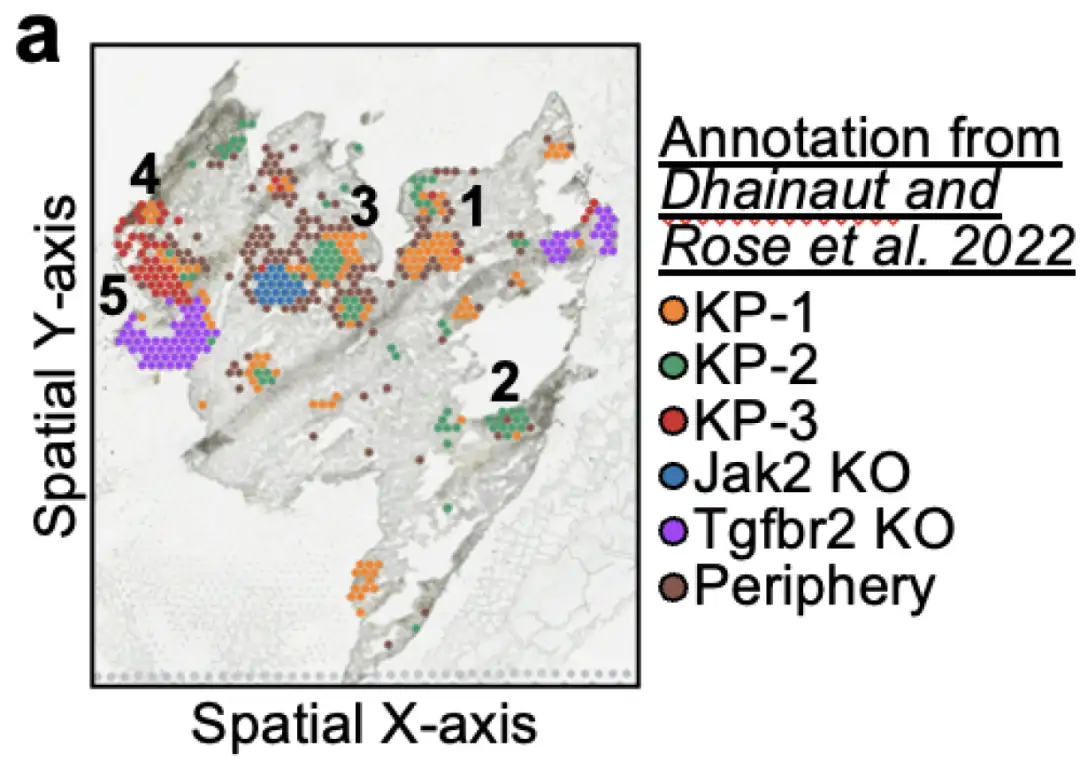
Um die Wirksamkeit von Celcomen bei der korrekten Erfassung von Störungseffekten in einem räumlichen Kontext zu demonstrieren, haben die Forscher es anhand eines In-vivo-Gesamttranskriptom-Datensatzes verglichen, der den Gen-Knockdown in der räumlichen Transkriptomik misst.Es heißt Perturbmap. Der Perturbmap-Datensatz enthält ein Mausmodell zur Untersuchung von KP-Lungenkrebs und darüber hinaus mögliche Knockouts von Jak2 oder Tgfbr2. Der Datensatz kommentiert 5 räumliche Regionen als Läsionsregionen, die Teile von 1) KP-Wildtyp-Krebs, oder 2) KP-Krebs mit Jak2-Knockout, oder 3) KP-Krebs mit Tgfbr2-Knockout sind, wie unten gezeigt:
Im Rahmen der Evaluierung der Fähigkeiten von CelcomenDer von den Forschern verwendete Datensatz zur fetalen Milz stammt aus https://developmental.cellatlas.io/fetalimmune,In log-normalisierter Form bereitgestellt, ist es klar, dass eine Log-Transformation und eine Normalisierung der Bibliotheksgröße durchgeführt wurden;Glioblastom-Datensatz von 10x Genomics,Es wurden die gleiche Normalisierung der Bibliotheksgröße, Counts per Million (CPM) und logarithmische Transformation zur Basis e durchgeführt; Darüber hinaus wurden nur Gene beibehalten, die in mindestens 100 Zellen exprimiert wurden.
Modellarchitektur: Ein neues Framework für die Kausalanalyse Celcomen
Das in dieser Studie vorgeschlagene Celcomen-Modell erreicht durch die Kombination von Lagrange-Mechanik und kausaler Inferenz eine Identifizierbarkeit der kausalen Inferenz und eine höhere Modellinterpretierbarkeit. Einfach ausgedrückt bedeutet Identifizierbarkeit, ob das Modell bei ausreichenden Daten und vernünftigen Annahmen kausale Zusammenhänge klar identifizieren kann, anstatt aufgrund mehrerer unterschiedlicher Annahmen oder Modelleinstellungen zu denselben Beobachtungsergebnissen zu führen – dies bietet einen neuen Rahmen für die Kausalanalyse in der räumlichen Transkriptomikforschung.
Celcomen basiert auf drei Kernannahmen: 1. Die erwartete Gen-Gen-Korrelation zwischen Nachbarn erster Ordnung muss genau mit den beobachteten Daten übereinstimmen; 2. Die erwartete Gen-Gen-Korrelation innerhalb desselben räumlichen Punkts/der gleichen Zelle muss genau mit den beobachteten Daten übereinstimmen. 3 Annahme der kausalen Suffizienz: Es gibt keine ungemessene gemeinsame Ursache zwischen den untersuchten Genpaaren.
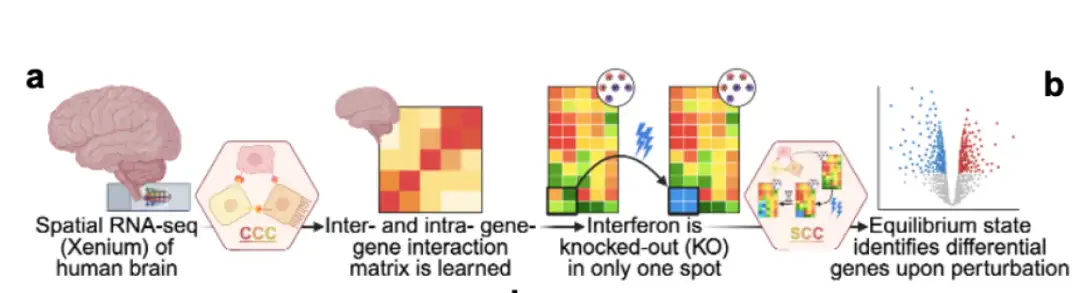
Wie in der folgenden Abbildung dargestellt:Celcomen ist in zwei Teile unterteilt: das Reasoning-Modul (CCE) und das Generation-Modul (SCE):
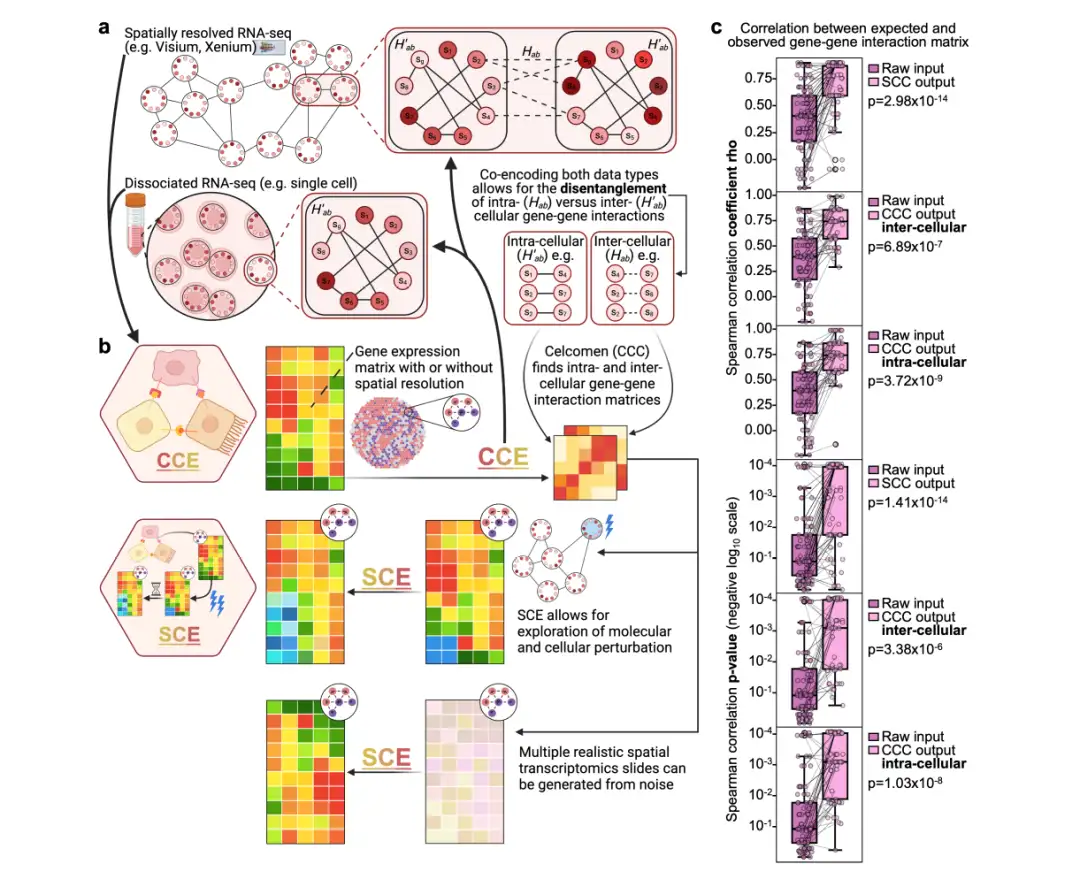
(a) Modul „Argumentation“ (CCE):Gen-Gen-Beziehungen können aus räumlich aufgelösten Transkriptomdaten (räumliche RNA-Sequenzdaten) und optional aus dissoziierten Einzelzell-RNA-Sequenzdaten (dissoziierte scRNA-Sequenzdaten) abgeleitet werden. Hervorgehobene Zell-Zell-Paare in räumlichen Daten und einzelne Zellen in Einzelzell-RNA-Sequenzdaten zeigen, wie CCE zwischen intrazellulären (H′ab) und interzellulären (Hab) Gen-Gen-Interaktionen unterscheiden kann.
b) Erzeugungsmodul (SCE):Die durch CCE erlernten Gen-Gen-Beziehungen werden verwendet, um kontrafaktisches Gewebeverhalten nach Zell- oder Genstörungen zu simulieren.
* Kontrafaktische Szenarien: Dies ist eine Methode zur Untersuchung des möglichen Verhaltens biologischen Gewebes unter verschiedenen hypothetischen Bedingungen, die hauptsächlich bei kausalen Schlussfolgerungen, Interventionssimulationen und biomedizinischer Modellierung verwendet wird. Dabei geht es darum, ein hypothetisches Szenario zu konstruieren, in dem beschrieben wird, wie sich das Verhalten eines biologischen Organismus von dem tatsächlich beobachteten Verhalten unterscheiden könnte, wenn ein Schlüsselfaktor verändert würde (z. B. Gen-Knockout, medikamentöse Intervention, Veränderung der äußeren Umgebung usw.).
Forschungsergebnisse: Das Celcomen-Modell ist bei der Entwirrung kausaler Zusammenhänge identifizierbar
Die Forscher überprüften die Identifizierbarkeit des Celcomen-Modells beim Erlernen kausaler Strukturen und beim Entwirren kausaler Beziehungen durch Experimente mit selbstkonsistenten synthetischen Daten und realen Daten.
Celcomen hat eine starke Selbstkonsistenz und Identifizierbarkeit
Wie in der folgenden Abbildung gezeigt, weist Celcomen im synthetischen Datensatz durchgängig eine starke Übereinstimmung zwischen den abgeleiteten Gen-Gen-Interaktionen und den realen Daten auf, was darauf hindeutet, dass Celcomen eine starke Selbstkonsistenz und damit Identifizierbarkeit aufweist.
* Selbstkonsistenz: In der Statistik, Optimierung und im maschinellen Lernen bedeutet Selbstkonsistenz normalerweise, dass die Annahmen, Ableitungen und Optimierungsprozesse des Modells zu einer stabilen Lösung konvergieren können.
* Identifizierbarkeit: bezieht sich darauf, ob die Modellparameter oder kausalen Effekte der Kausalbeziehung basierend auf den beobachteten Daten im Kausalinferenzmodell eindeutig bestimmt werden können.
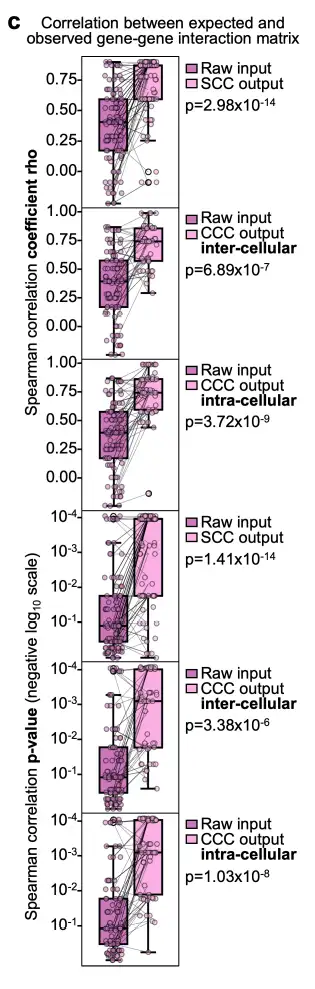
Die Forscher bestätigten außerdem die Identifizierbarkeitsgarantien des Celcomen-Modells anhand realer menschlicher Daten, indem sie es auf räumliche Transkriptomabschnitte mehrerer menschlicher fetaler Milzen anwendeten und Spearman-Korrelationskoeffizienten zwischen den beiden Gen-Gen-Interaktionsmatrizen im Bereich von 0,5 bis 0,6 beobachteten. Darüber hinaus sind die erfassten genetischen Interaktionen in intra- und interzellulären Matrizen biologisch plausibel, da sie bekannten biologischen intra- und interzellulären Prozessen folgen.
Dies belegt die Identifizierbarkeit von Celcomen und bestätigt seine implizite Stabilität und Robustheit, die über theoretische und synthetische Daten hinausgeht und auch in echten menschlichen Proben beobachtet werden kann.
Fähigkeit zur kausalen Entkopplung: Celcomen kann die Quellen intrinsischer und extrinsischer Transkriptomvariationen erfolgreich entwirren
Anschließend testeten die Forscher die Fähigkeit von Celcomen, Genregulationsprogramme innerhalb und zwischen Zellen zu entwirren (Entkopplungsfähigkeit). Sie wendeten Celcomen in einer realen klinischen Umgebung am Menschen an, um einen räumlichen Transkriptom-Datensatz mit Einzelzellauflösung eines menschlichen Glioblastoms (Hirnkrebs) zu analysieren, wie in der folgenden Abbildung dargestellt. Die Forscher stellten fest, dass Celcomen in der Lage war, die Quellen intrinsischer und extrinsischer Transkriptomvariationen erfolgreich aufzudecken.
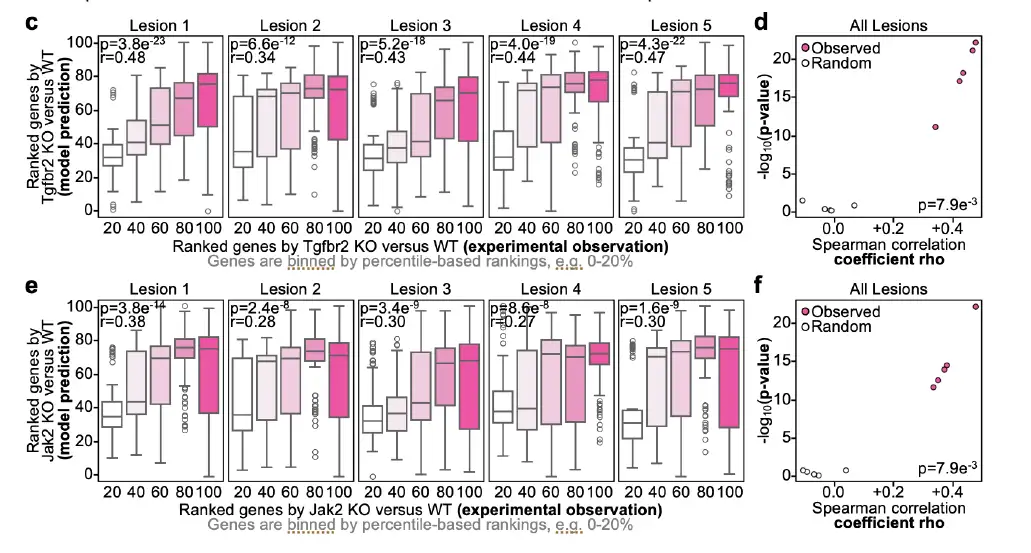
Räumliche kontrafaktische Validierung in vivo: Celcomen schneidet deutlich besser ab als eine zufällige Baseline
Um die Wirksamkeit von Celcomen weiter nachzuweisen, führten die Forscher einen Benchmarktest mit dem In-vivo-Gesamttranskriptom-Datensatz Perturbmap durch. Die Ergebnisse zeigten, dass die Spearman-Korrelation zwischen Vorhersagen und In-vivo-Messungen für alle Läsionen zwischen 0,28 und 0,47 lag. Um die Bedeutung dieser Leistung zu beurteilen, verglichen die Forscher das Modell mit einer zufälligen Basislinie, wobei Celcomen mit zufällig gemischten Daten ausgeführt wurde. Die Ergebnisse zeigen, dass Celcomen mit einem p-Wert von 0,0079 signifikant besser abschneidet als die zufällige Basislinie, wie in der folgenden Abbildung (cf) dargestellt:
Zusammenfassend eröffnet das in dieser Studie vorgeschlagene Modell einen neuen Weg zur Erzielung mechanistischer Erklärbarkeit durch kausale Inferenz. Wie in Experimenten gezeigt wurde, können die Forscher dank der kausalen Identifizierbarkeit des Celcomen-Modells die Parameterwerte des neuronalen Netzwerks mit hoher Genauigkeit wiederherstellen. Die Fortschritte von Celcomen haben erhebliche Auswirkungen auf den biomedizinischen Bereich, indem sie beispielsweise aufdecken, wie Krankheiten zu Gewebeversagen führen, und überprüfbare Hypothesen über den Nutzen von Behandlungen ermöglichen. Der Wert von Celcomen wird mit dem technologischen Fortschritt weiter steigen und zu Verbesserungen bei der Krankheitsmodellierung und dem mechanistischen Verständnis führen.
Künstliche Intelligenz erschließt das Potenzial der räumlichen Transkriptomik
Die in dieser Studie erzielten relevanten Ergebnisse stellen eine weitere Weiterentwicklung der räumlichen Transkriptomik dar – die Technologie der räumlichen Transkriptomik ist einer der größten Durchbrüche auf dem Gebiet der Bioinformatik der letzten Jahre. Diese Technologie hat das Paradigma der biomedizinischen Forschung grundlegend verändert, indem sie detaillierte, räumlich lokalisierte molekulare Merkmale liefert und es Bioforschern ermöglicht, die Struktur und Funktion von Gewebe mit einer beispiellosen Auflösung aufzuklären.
In den letzten Jahren hat sich die Technologie der räumlichen Transkriptomik rasant weiterentwickelt und es wurden kontinuierlich Daten gesammelt. Auf dieser Grundlage wies der im August 2024 veröffentlichte Artikel „Nature Methods Special Issue Comment: Using the „Key“ of Artificial Intelligence to Open the „Lock“ of Spatial Omics“ darauf hin, dassKünstliche Intelligenz hat das Potenzial, das volle Potenzial der räumlichen Omics freizusetzen, die Integration komplexer Datensätze zu erleichtern und neue biomedizinische Erkenntnisse zu gewinnen.
Insbesondere kann KI die Integration von räumlicher Transkriptomik und scRNA-Sequenzierung erleichtern, sodass Forscher transkriptomweite räumliche Genexpressionsprofile auf Einzelzellebene messen können. Darüber hinaus kann KI durch die Integration räumlicher Omics- und histologischer Bilddaten hochauflösende, umfassende dreidimensionale räumliche Gewebekarten erstellen, die ein breites Spektrum an Omics-Modalitäten abdecken. Da die Anzahl verfügbarer Datensätze wächst, können multimodale Large Language Models (MM-LLMs) anhand räumlicher Omics-, medizinischer Bildgebungs- und klinischer Textdaten für Aufgaben in der biomedizinischen Forschung und Präzisionsmedizin trainiert werden.
Oktober 2023Die Forschungsgruppe von Zhang Shihua am Institut für Mathematik und Systemwissenschaften der Chinesischen Akademie der Wissenschaften hat einen Artikel in Nature Computational Science veröffentlicht.Veröffentlichung eines Forschungspapiers mit dem Titel „Integration räumlicher Transkriptomikdaten unter verschiedenen Bedingungen, Technologien und Entwicklungsstadien“. Diese Arbeit führte ein neues integriertes Analysetool, STAligner, für räumliche Transkriptomdaten mehrerer Scheiben biologischen Gewebes aus unterschiedlichen Technologien, zu unterschiedlichen Entwicklungszeitpunkten und bei unterschiedlichen Krankheitszuständen ein. Es kann Forschern helfen, bei der Durchführung räumlicher Transkriptomikanalysen wichtige neue biologische Erkenntnisse zu gewinnen.
*Originalpapier:
https://www.biorxiv.org/content/10.1101/2022.12.26.521888v1.full.pdf
Um die vielschichtigen Herausforderungen der räumlichen Transkriptomdatenanalyse zu lösen, werden im Juli 2024Forschungsgruppe von Associate Professor Zhang Qiangfeng an der School of Life Sciences, Tsinghua University/Structural Biology Advanced Innovation Center/Tsinghua-Peking University Joint Center for Life Sciences,Ein Forschungspapier mit dem Titel „Tissue module discovery in single-cell resolution spatial transcriptomics data via cell-cell interaction-aware cell embedding“ (Entdeckung von Gewebemodulen in räumlichen Transkriptomikdaten mit Einzelzellauflösung über die Einbettung von Zellen zwischen Zellen) wurde online in der Zeitschrift Cell Systems veröffentlicht. In dieser Studie wurde ein künstlicher Intelligenzalgorithmus SPACE (Spatial Transcriptomics Data Analysis via „interaction-aware“ Cell Embedding) entwickelt, der auf dem Graph Autoencoder Deep Learning Framework basiert und räumliche Zelltypen identifizieren und Gewebemodule aus räumlichen Transkriptomdaten mit Einzelzellauflösung entdecken kann und für groß angelegte räumliche Transkriptomforschung verwendet werden kann.
Mit Blick auf die Zukunft wird erwartet, dass Forscher durch die Nutzung der leistungsstarken Rechenleistung und der Deep-Learning-Algorithmen der KI eine neue Dimension der räumlichen Transkriptomik erschließen, die Effizienz der Krankheitsforschung, der Arzneimittelentwicklung und der personalisierten Medizin deutlich verbessern und es Wissenschaftlern ermöglichen, die räumliche Heterogenität biologischer Systeme mit beispielloser Präzision zu untersuchen und so bahnbrechende wissenschaftliche Entdeckungen zu machen.
Quellen:
1.https://openreview.net/forum?id=Tqdsruwyac
2.https://www.thepaper.cn/newsDetail_forward_28521641
3.https://www.cas.cn/syky/202310/t20231020_4981872.shtml








