Command Palette
Search for a command to run...
LAMMPS 入门教程:npt 控温估计 FCC Cu 熔点
LAMMPS 全称为 Large-scale Atomic/Molecular Massively Parallel Simulator,是一种经典的分子动力学仿真代码,专注于材料建模。它旨在在并行计算机上高效运行,并且易于扩展和修改。 LAMMPS 最初由美国能源部机构桑迪亚国家实验室开发,现在包括来自许多机构的许多研究小组和个人的贡献。
本教程使用 LAMMPS CPU 即可运行。
通过本教程的学习,您将能够:
* 理解 npt 控温操作流程
* 使用 dump 和 fix 指令将数据预处理
效果示例

一、运行前准备
在这里我们首先介绍一些需要使用的输入文件、势函数类型和 npt 控温流程。
1. 输入文件
进入 ./melt_u3,melt.in 文件如下:
units real
dimension 3
boundary p p p
atom_style atomic
# 读取铜结构
read_data cu
#选用 eam potential
pair_style eam
pair_coeff * * Cu_u3.eam
#每隔 100 时间步长于屏幕输出
thermo 100
thermo_style custom step temp pe press vol lx density
#将原子轨迹输出到文件 md.lammpstrj
dump 2 all custom 100 md.lammpstrj id type x y z
#结构优化
minimize 1.0e-10 1.0e-10 10000 10000
#nvt 压强弛豫
fix 1 all nvt temp 1000 1000 100
run 10000
unfix 1
#npt 体积弛豫
fix 1 all npt temp 1000 1000 100 iso 1 1 1000
run 10000
unfix 1
#将温度和体积量分别存储到 t 和 v 中
variable t equal "temp"
variable v equal "vol"
#npt 控温由 1000K 升温至 2000K 。
#升温速率为 (2000-1000)/100000=0.001 K/fs=1K/ps
fix 1 all npt temp 1000 2000 100 iso 1 1 1000
fix 2 all ave/time 100 10 10000 v_t v_v file t_v.txt
run 1000000
2. 势函数类型
该项目提供了 eam 势函数的演示模型
eam
./melt_u3
对应的势函数模块
pair_style eam
pair_coeff * * Cu_u3.eam
3.npt 控温流程
主要流程为:
nvt 压强弛豫,npt 体积弛豫
#结构优化
minimize 1.0e-10 1.0e-10 10000 10000
#nvt 压强弛豫
fix 1 all nvt temp 1000 1000 100
run 10000
unfix 1
#npt 体积弛豫
fix 1 all npt temp 1000 1000 100 iso 1 1 1000
run 10000
unfix 1
npt 升温
#npt 控温由 1000K 升温至 2000K 。
#升温速率为 (2000-1000)/100000=0.001 K/fs=1K/ps
fix 1 all npt temp 1000 2000 100 iso 1 1 1000
fix 2 all ave/time 100 10 10000 v_t v_v file t_v.txt
run 1000000
二、实操运行步骤(以 CPU 为例)
1. 创建并启动容器分子动力学模拟
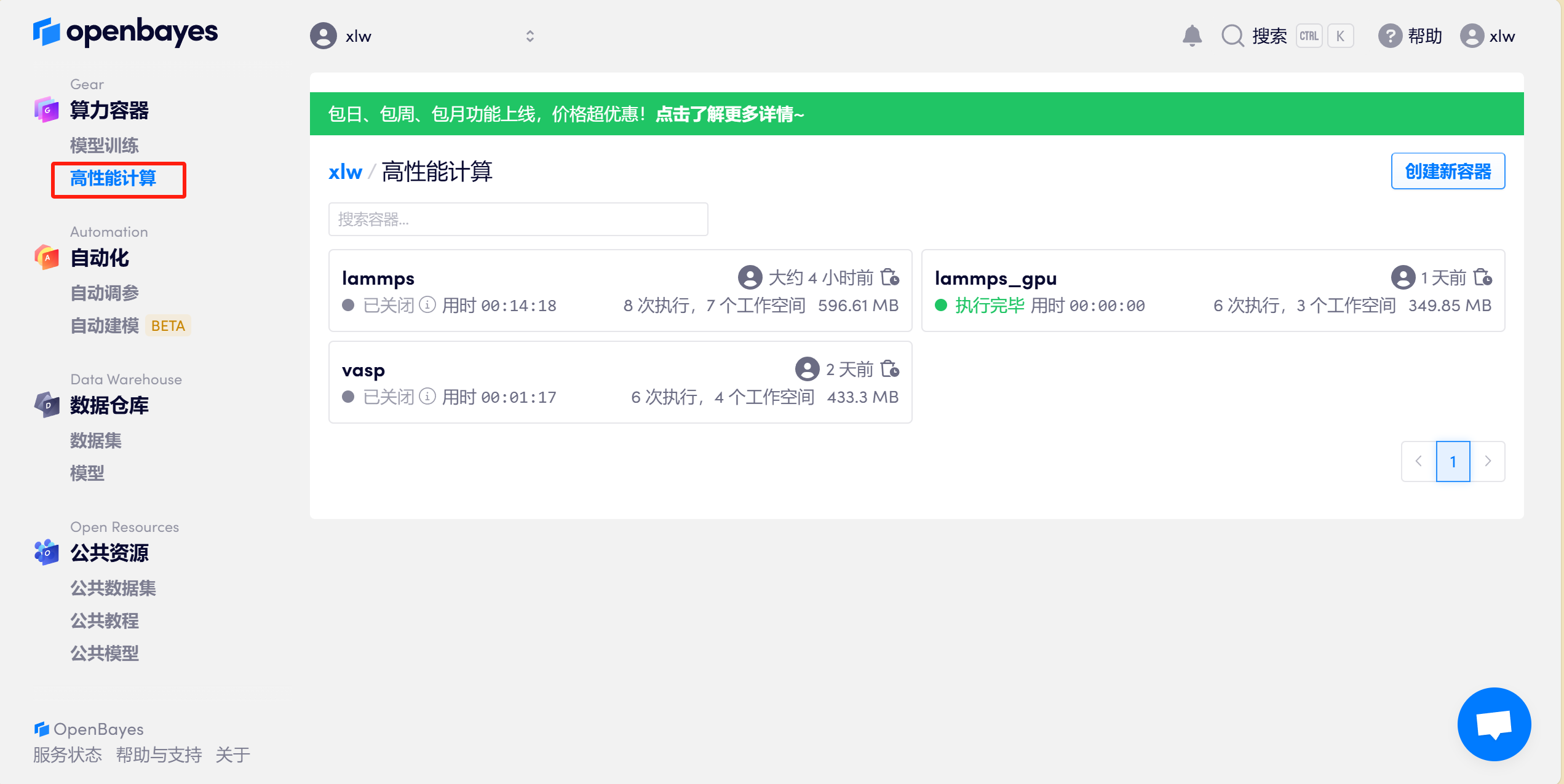
1.1 打开 OpenBayes 个人用户界面,点击左侧「高性能计算」> 右上角「创建新容器」
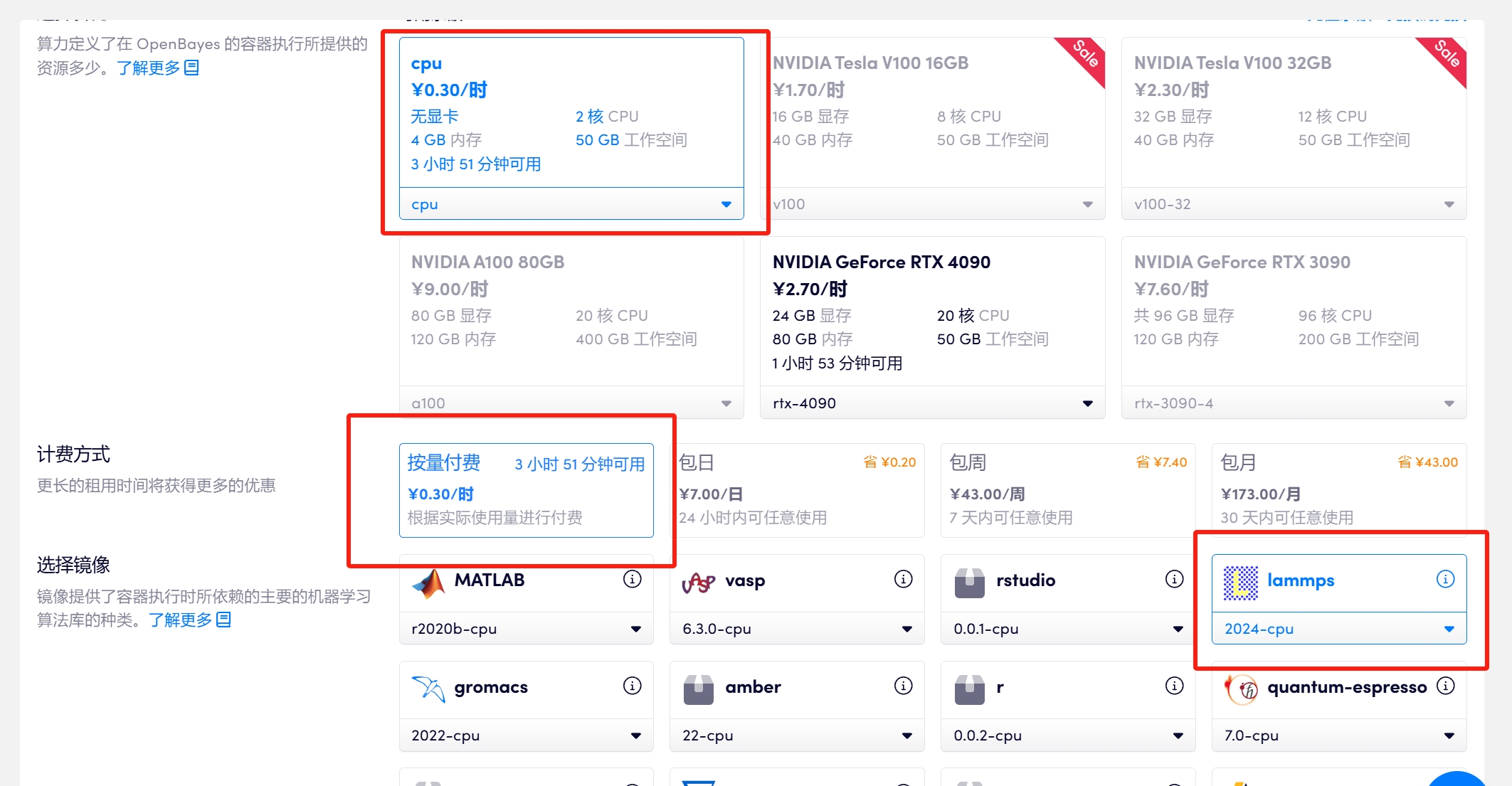
1.2 算力选择「CPU」>「lammps-2024-cpu」取一个容器名称,例如 lammps> 选择「工作空间」,然后点击「下一步:选择算力」
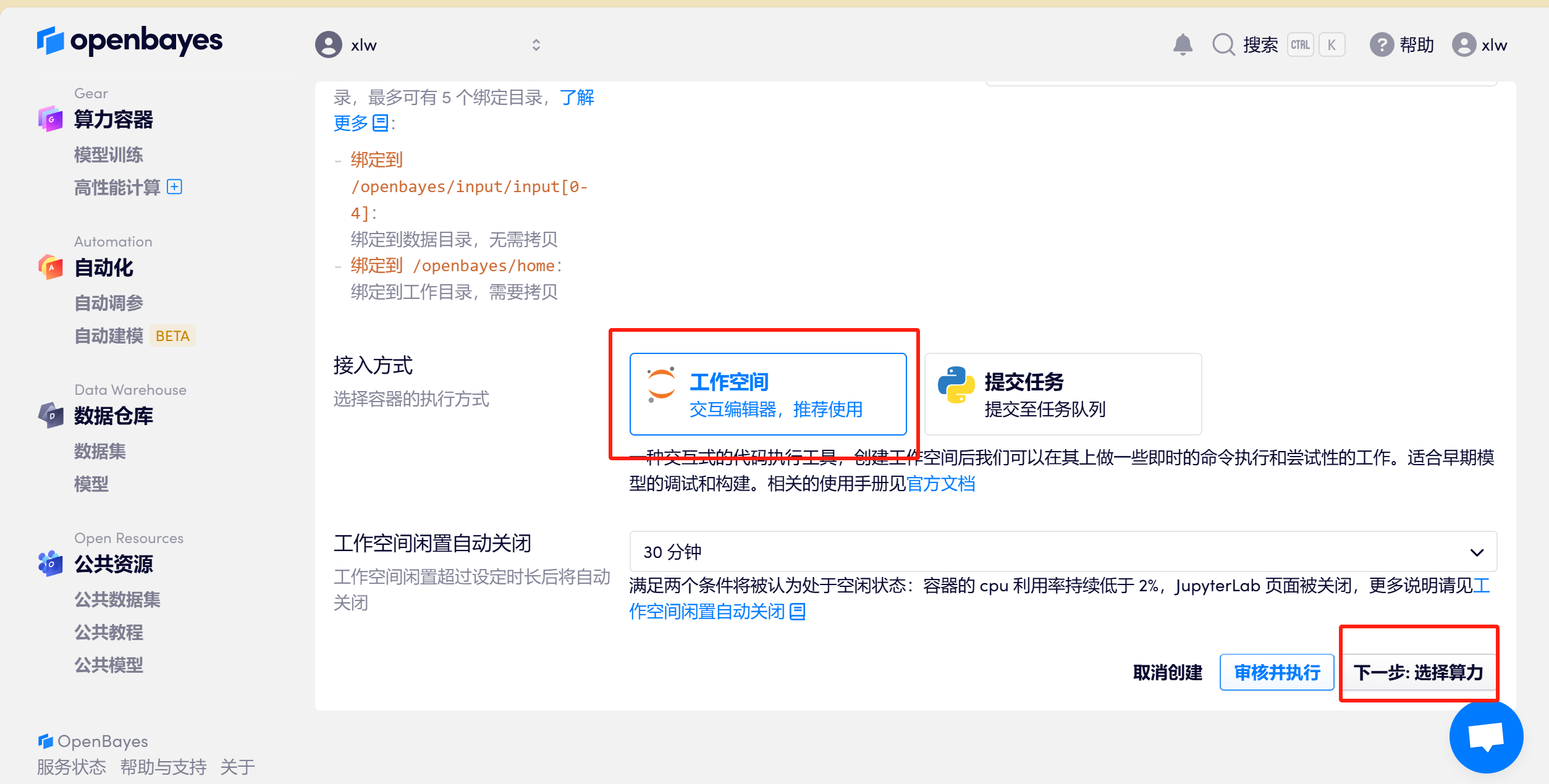
1.3 取一个容器名称,例如 lammps> 选择「工作空间」,然后点击「执行」
2. 进行分子动力学模拟
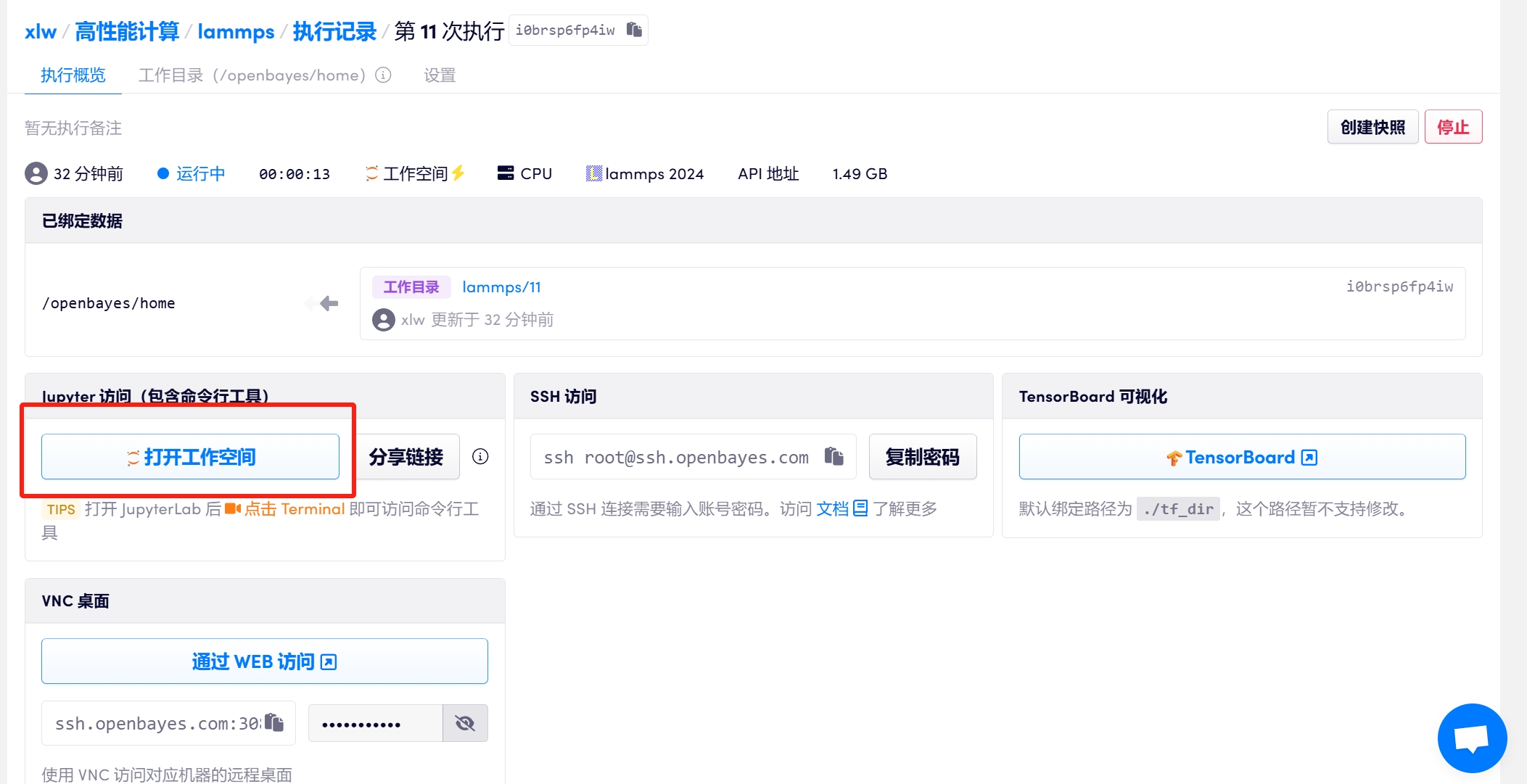
2.1 待容器成功运行后,点击左侧打开工作空间
2.2 将压缩包拖到左侧文件区上传
2.3 然后打开终端
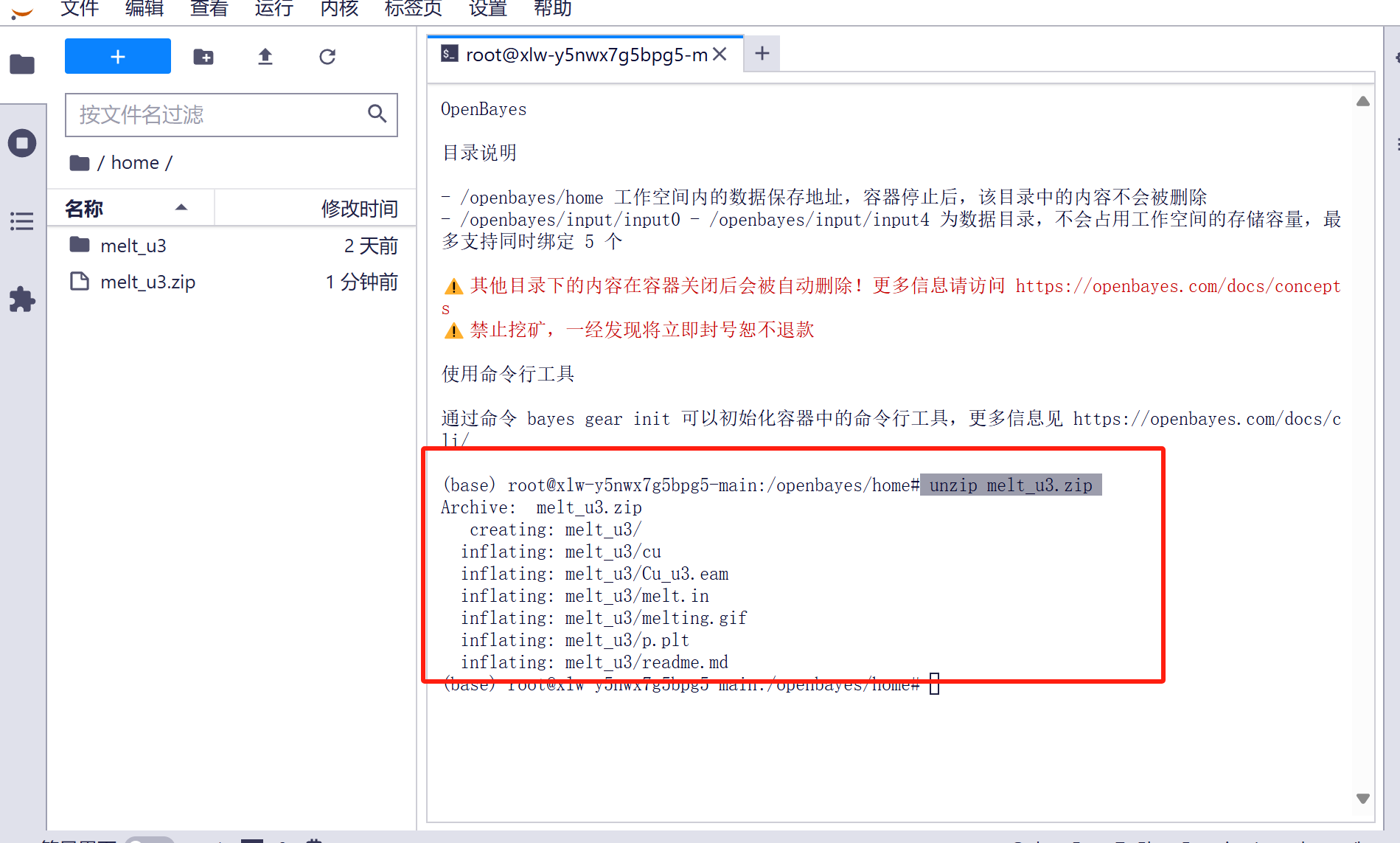
2.4 解压(本容器已将压缩包解压完毕,可以跳过此步骤):输入 unzip melt_u3.zip 解压压缩包
unzip melt_u3.zip
Archive: melt_u3.zip
creating: melt_u3/
inflating: melt_u3/cu
inflating: melt_u3/Cu_u3.eam
inflating: melt_u3/melt.in
inflating: melt_u3/melting.gif
inflating: melt_u3/p.plt
inflating: melt_u3/readme.md
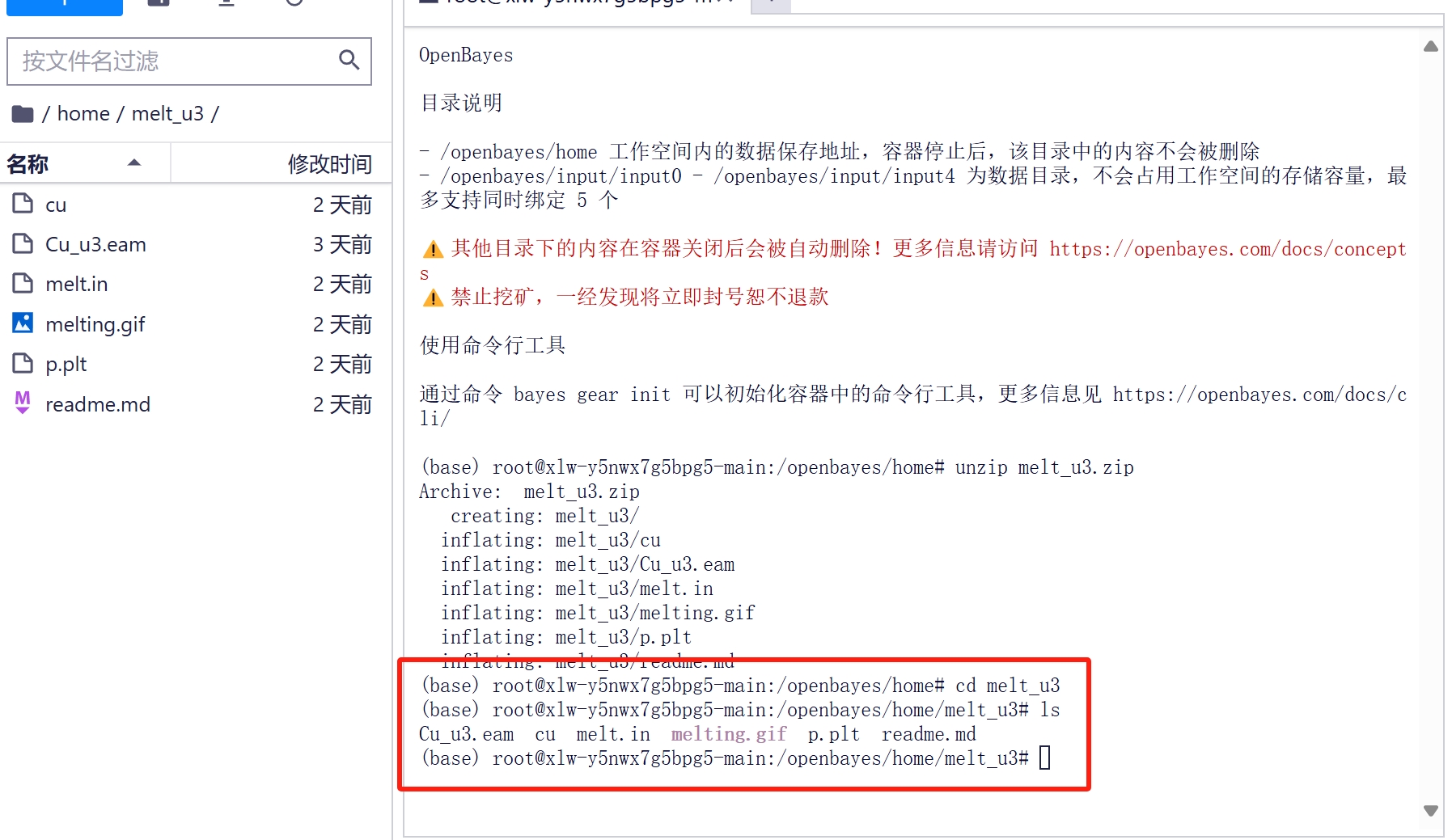
2.5 输入 cd melt_u3 进入解压目录,使用 ls 查看文件
ls
Cu_u3.eam cu melt.in melting.gif p.plt readme.md
2.6 运行 lammps
mpirun -np 2 lmp < melt.in | tee out
2.7 屏幕输出会同步在 out 文件中,总运行时间大约需要 5 分钟
2.8 运行完毕后可在文件夹中得到 t_v.txt 等输出文件
2.9 t_v.txt 文件由输入文件中 fix 命令控制输出
#将温度于体积量分别存储到 t 和 v 中
variable t equal "temp"
variable v equal "vol"
fix 2 all ave/time 100 10 10000 v_t v_v file t_v.txt
生成
3. 数据处理
3.1 更新 apt 源
apt-get update --fix-missing
3.2 输入 apt install gnuplot 安装 gnuplot 工具,并输入 y 回车确认
apt install gnuplot
3.3 运行命令 gnuplot p.plt 可得到 t-v 图
gnuplot p.plt
3.4 双击左侧 t-v 图可得到估计的 Cu 熔点
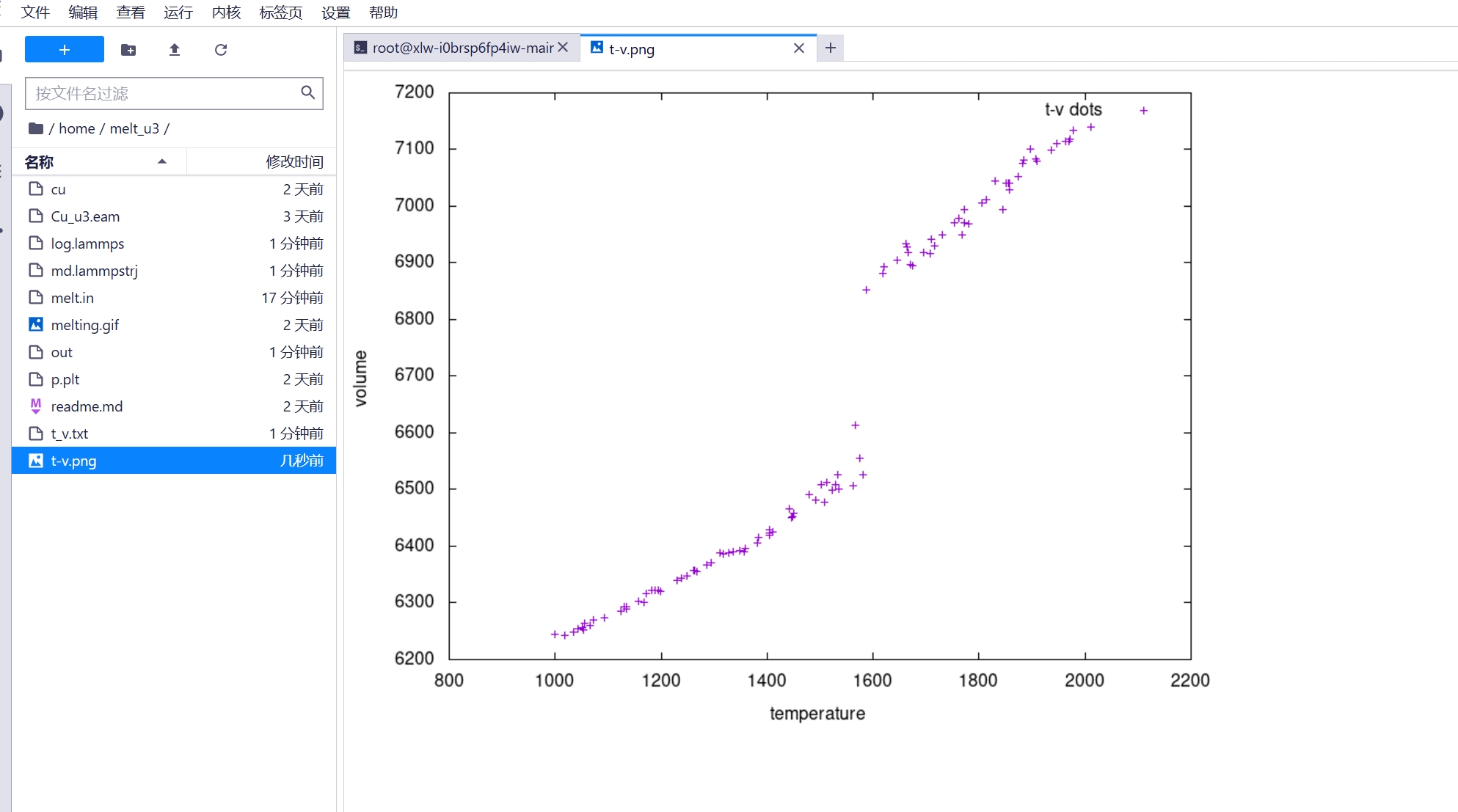
可以看到熔点大约在 1600K 附近,与实际 1357.77K 有差距,这是由于升温过快,铜还来不及融化导致的。
升温过程原子轨迹由
#将原子轨迹输出到文件 md.lammpstrj
dump 2 all custom 1 md.lammpstrj id type x y z
存储至 md.lammpstrj 中,我们将其下载下来
3.5 可用 ovito 进行可视化