Command Palette
Search for a command to run...
多模态模型加速新材料与工业应用匹配,无需完整晶体结构即可预测材料性质
橘生淮南则为橘,橘生淮北则为枳。同一颗种子落在不同的土壤和气候中收获的果实也大相径庭,这一自然界的成长规律在材料化学领域同样凸显——新材料面向不同的应用场景所激发的性能也不尽相同。有研究表明,科学家每年都会创建出数十万种新材料,它们就像无数颗潜力巨大的「种子」,需要植根于与之匹配的环境中才能茁壮成长。
尽管目前的新材料设计往往是针对特定应用而合成的,但是其在不同领域往往也有潜在用途,而如何快速确定新材料的应用场景仍是一项颇具挑战的任务。以应用广泛的晶体材料金属有机框架(MOFs)为例,其最显著的用途是氢气、甲烷等气体的储存介质,并在膜、薄膜器件、催化和生物医学成像等领域展现出优异的性能潜质。而确定 MOFs 最优应用的传统途径均需依赖材料特性作为中介判断依据,但测试成本高(时间、设备、专业知识等)。此外,对于计算筛选与机器学习方法而言,则需要基于完整晶体结构来预测特性,但晶体结构解析耗时且在 MOFs 合成后无法即时获取。
针对于此,加拿大多伦多大学化学工程与应用化学系的研究团队提出了一种基于多模态机器学习模型的新方法,利用 MOFs 合成后即可获得的信息来预测其潜在性能和用途,例如其粉末 X 射线衍射图谱(PXRD)以及所使用的合成化学物质。研究团队为模型添加了应用推荐系统,能在 MOFs 合成后的第一时间给出应用建议,该研究加速了金属有机框架(MOFs)的合成与应用场景的连接。
相关研究以「Connecting metal-organic framework synthesis to applications using multimodal machine learning」为题,发表于 Nature Communications 。
研究亮点:
* 该方法仅使用合成后即可获得的信息来预测 MOFs 的潜在性质和用途,在 MOFs 合成后的第一时间进行应用推荐,大幅缩短材料从合成到应用的周期;
* 模型预测性能与需要精确晶体结构输入的先进模型(如 CGCNN 、 MOFormer)相当,且在部分条件下表现更优,在面对实验噪声、晶体结构缺陷等情况下依旧稳定可靠,具有良好的鲁棒性;
* 该研究结合可视化应用推荐系统构建了一个合成-预测-应用闭环系统;
论文地址:
https://www.nature.com/articles/s41467-025-60796-0
关注公众号,后台回复「MOFs」获取完整 PDF
更多 AI 前沿论文: https://hyper.ai/papers
「数据即合成现场」:面向应用预测的 MOFs 数据构建策略
在本次研究中共使用了 6 个金属有机框架(MOFs)数据库进行模型训练与评估:CoRE-2019 、 BW20K 、 ARABG 、 QMOF 、 hMOF 、 CSD 子集。其中:
* hMOF 提供超大规模的假想结构库,有助于提高模型泛化能力。
* BW20K 和 ARABG 用于增强多样性和支持小样本任务。
* CSD 子集用于测试模型在含有实验偏差下的鲁棒性。
研究团队利用 CoRE 2019 、 BW20K 、 ARABG 、 QMOF 、 hMOF 数据库中提供的晶体结构,通过 pymatgen 的 XRD 模块计算生成 0 到 90 度的模拟 PXRD 图谱,以模拟实际实验中合成后即能获得的结构表征信息。化学前驱体信息则由金属节点与有机连接体组成,构建格式为:[金属类型].[有机连接体],以此输入到模型的 Transformer 通道并进行 分词处理。
自监督预训练驱动的多模态学习框架
研究团队提出了一种自监督预训练驱动的多模态学习框架,旨在摆脱对完整晶体结构的依赖,仅利用合成后即可获得的信息来预测 MOF 的性质与应用潜力。
这一自监督多模态模型的工作流程如下图所示,以前驱体字符串和粉末 X 射线衍射(PXRD)谱作为输入,分别通过 Transformer 和卷积神经网络(CNN)进行嵌入,并传递给回归头(regression head)进行微调。其中前驱体提供了有关材料化学性质的信息,而 PXRD 图谱则补充了有关整体几何结构的信息。
通过 Transformer 编码的化学前驱体字符串,以及 CNN 处理的 PXRD 光谱,经特征拼接与投影构建成统一的表征空间。为了弥补「前驱体+PXRD」无法直接表征局部化学环境的不足,研究团队引入了自监督预训练机制,将模型输出与晶体图卷积神经网络(CGCNN)的嵌入进行对齐,并通过 Barlow Twins 损失约束互相关矩阵接近单位矩阵,从而引导模型习得局部化学环境的表达能力。
在此基础上,经过大规模无标签数据的自监督训练,模型能够在有限标注样本下快速收敛,并实现对孔隙结构、化学依赖特性以及量子化学性质的高精度预测。
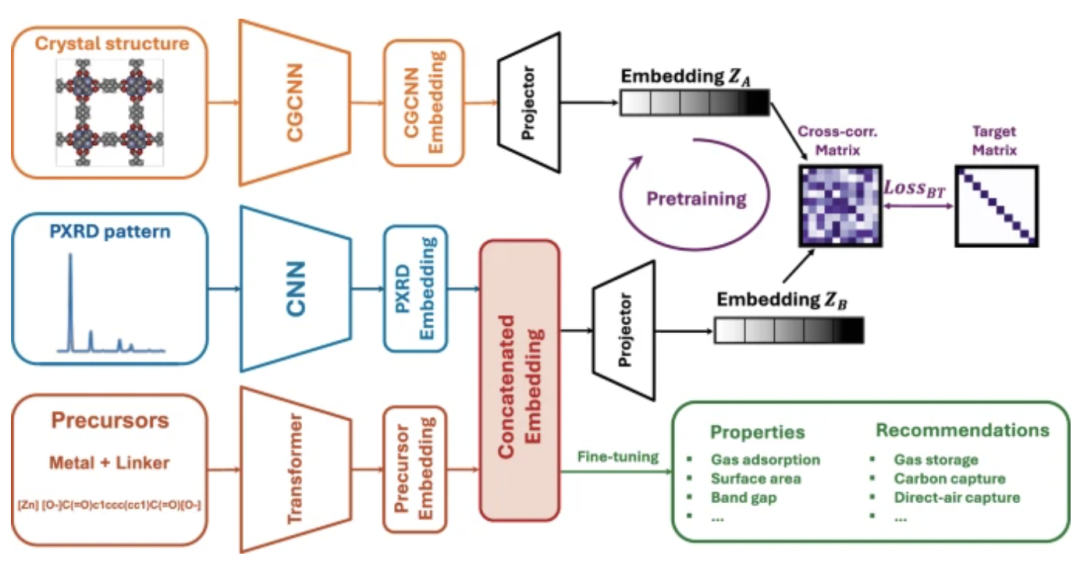
具体而言,基于从 MOFs 数据库中获取的晶体结构,即使在数据量较小的情况下,该方法也能准确预测各种特性,包括孔隙结构、化学依赖特性和量子化学特性。
在自监督与训练的环节通过构建自监督学习(SSL)管道,在晶体图卷积神经网络(CGCNN)和模型之间进行表征学习,避免模型无法从输入中理解 MOF 局部环境的局限性。模型的权重得以初始化,使其能够快速收敛到解决方案。针对 CGCNN 嵌入进行自监督学习,从 CGCNN 和模型的投影器中提取每个大小为 512 的嵌入,构建一个形状为(512,512)的互相关矩阵,使用巴洛双胞胎损失函数(Barlow-Twin loss)来最小化差异,使互相关矩阵接近单位矩阵,从而实现表征学习。
多模态模型的各项评估
为了证明模型可以有效预测各种 MOFs 特性,为 MOFs 合成与应用相结合奠定基础,研究团队对模型的准确性进行了评估,用斯皮尔曼等级相关系数(SRCC)和平均绝对误差(MAE),评估模型在几何依赖性能、化学依赖性能和量子化学性能上的预测精度,并与 CGCNN 、 MOFormer 和基于描述符的机器学习模型进行基准对比。
结果显示,该模型的模型精度与依赖完整晶体结构的模型相当,甚至在几何性能上优于 CGCNN 和 MOFormer,从而验证了仅用合成信息即可实现高准确率的性质预测,为 MOFs 合成到应用的快速匹配奠定实验基础。
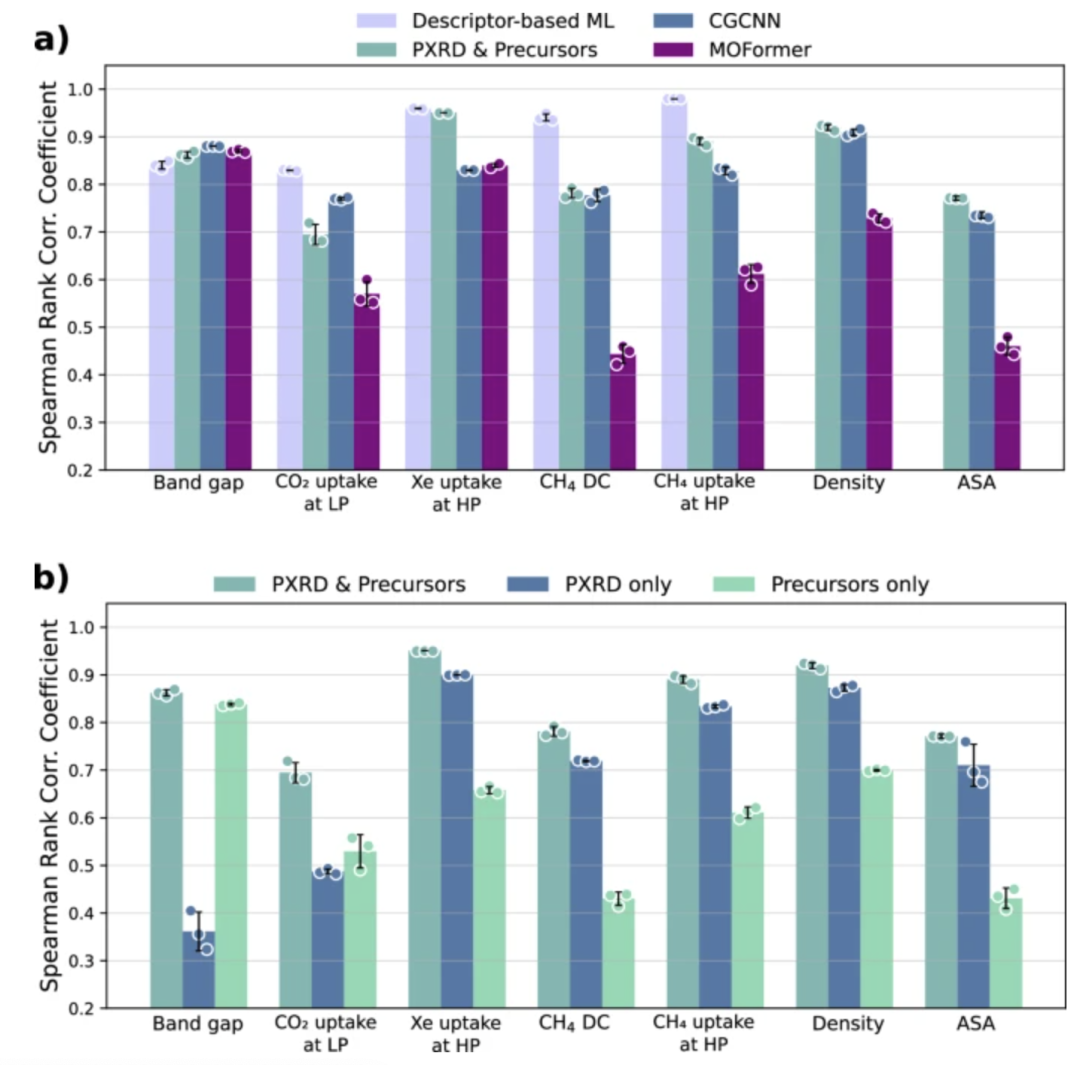
除此之外团队进行了消融实验,将仅依赖化学前驱体的模型以及仅依赖 PXRD 的模型,与本研究的多模态模型进行比较。结果表明,仅接受化学前体作为输入的模型无法有效捕捉 MOF 的整体结构,在几何相关和纯几何性质方面得分较低;而仅接受 PXRD 的模型虽能很好地捕捉 MOF 的整体结构,但无法反映局部环境,在化学相关和量子化学性质(如低压下的 CO₂ 吸附和带隙)方面得分较低,二者均存在一定的短板。结果表明只有将 PXRD(提供几何信息)与前体字符串(提供化学信息)结合,多模态模型才能在三类性质预测中全面准确,单独使用任一模态则表现明显不足。
模型稳定性验证:应对结构误差与实验噪声的鲁棒性评估
稳定性是评估机器学习模型能否在实际场景中可靠应用的重要指标。为此,研究团队系统性地评估了所提出的多模态模型在非理想条件下的鲁棒性。首先,研究人员利用从剑桥结构数据库(CSD)提取的实验晶体结构,计算出相应的 PXRD 图谱,用以模拟真实实验中常见的结构偏差,例如缺失氢原子、存在结合或未结合的溶剂等,将评估聚焦于一个几何相关属性:用于甲烷储存应用的高压下甲烷吸附能力预测。
结果表明,模型在上述变异条件下依然能够保持良好的预测能力,对 CH₄ 高压吸附性能的排序具有较强的一致性,相对误差控制在 13% 以下,展现出较高的鲁棒性。
在此基础上,团队进一步引入实际测量的 PXRD 图谱进行测试,验证模型在面对仪器噪声、温度波动等实际测量误差时的稳定性。尽管部分样本中模拟与实验图谱存在显著差异,模型在大多数情况下依然给出了与模拟图谱相近的推荐结果,仅在噪声显著或峰位错位明显的个别案例中出现差异。结合以上实验表明,该多模态模型不仅在理想结构输入条件下具有高度预测准确性,而且在实验结构不完善或 PXRD 存在噪声的情况下,仍保持稳健性能,验证了其在实际材料研究和应用中的广泛适用性。
下图展示了模型推荐结果,比较了模拟 PXRD 图谱和实验 PXRD 图谱的差异:
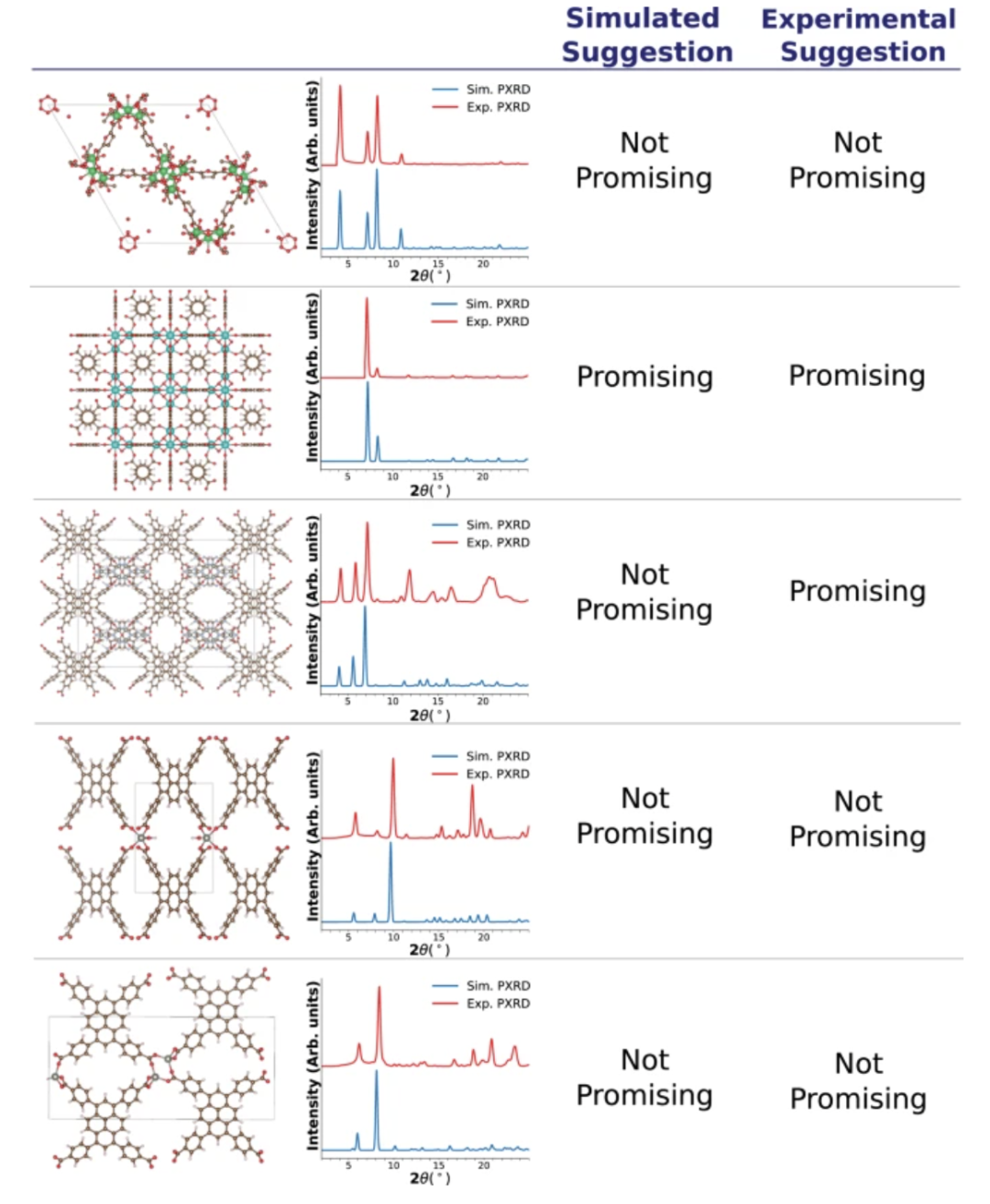
合成-应用一体化推荐系统
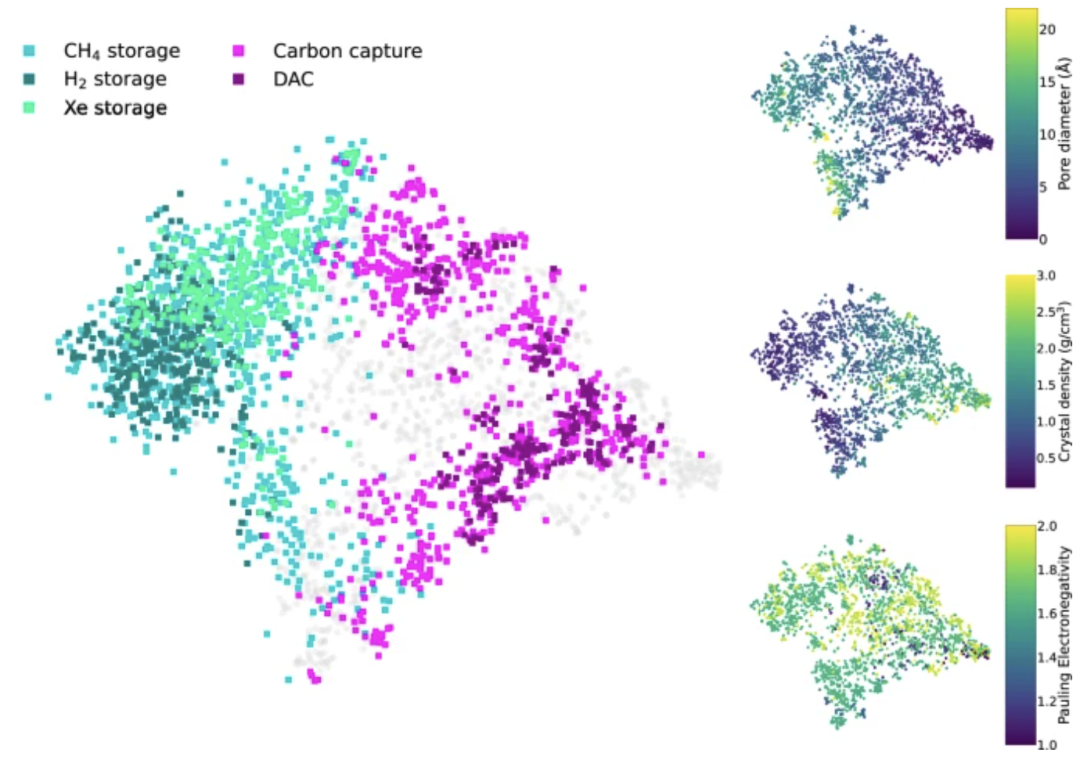
基于模型的出色表现,研究人员构建了一个可视化的潜在应用推荐系统,能够根据预测的材料性能将新合成的 MOFs 与潜在应用(如气体储存、碳捕获等)进行匹配。其使用 t-SNE 技术获取模态模型潜在空间的投影,用颜色表示金属有机骨架 (MOFs) 的推荐应用。下图展示了将合成信息映射至应用场景:
为了验证模型对未来材料应用的预测能力,研究者们进行了一项时间回溯实验(time-travel experiment)。使用 2017 年之前存储在 CSD 数据库中的 CoRE-2019 条目训练模型,并使用 2017 年之后存储的条目作为测试集,以此模拟对未来材料的预测。实验的目标是预测这些 MOFs 在二氧化碳吸附这一特定应用中的表现,结果显示,模型成功地识别出了 18 种有潜力用于碳捕获的 MOF,这 18 种 MOF 中,有 15 种原本是为其他应用而设计的。
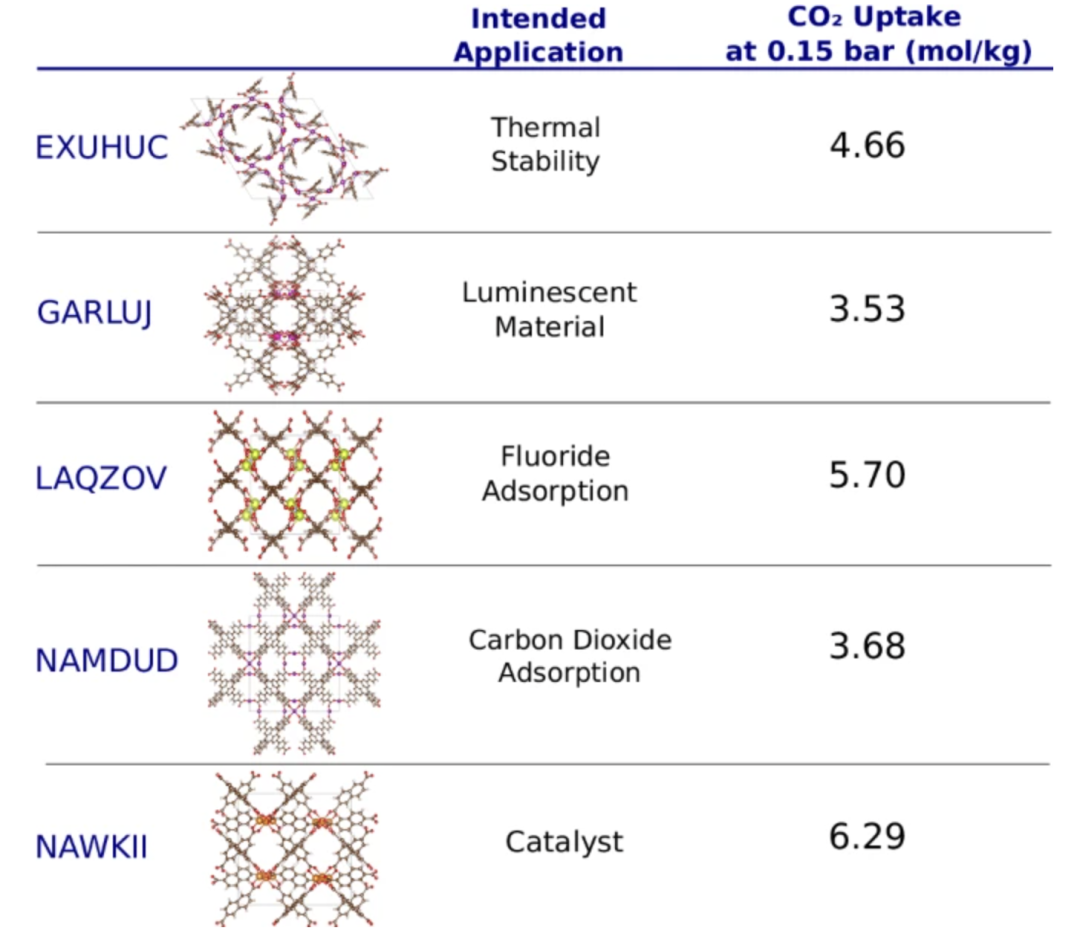
其中一些 MOFs 及其基于相应的预期合成应用
机器学习助力材料科学领域革命
本文介绍了一种无需晶体结构即可准确预测 MOFs 多种性质并匹配应用的多模态机器学习方法,而这种由数据驱动的浪潮,正在不同时空朝着更广泛的材料体系蔓延。例如,北京科技大学谢建新&宿彦京团队针对可解释机器学习在材料科学中的应用进行了探讨。指出了通过将材料知识与机器学习相结合,可以显著提高模型的泛化能力和预测精度,为材料科学领域的发展打开新视角。相关研究以「Interpretable Machine Learning Applications: A Promising Prospect of AI for Materials」为题,发表于 Advanced Functional Materials 。
论文地址:
https://advanced.onlinelibrary.wiley.com/doi/abs/10.1002/adfm.202507734
来自美国阿贡国家实验室的研究团队,提出了一种生成式 AI 框架 GHP-MOFsassemble,该框架能够随机生成并组装新的 MOFs 结构,通过分子动力学模拟筛选高稳定性的 MOFs 结构,并用晶体图神经网络 (Crystal Graph Convolutional Neural Network, CGCNN) 和 Grand Canonical Monte Carlo simulations 来测试 MOFs 对二氧化碳的吸附能力。相关研究以「A generative artificial intelligence framework based on a molecular diffusion model for the design of metal-organic frameworks for carbon capture」为题,发表于 Communications Chemistry 。
论文地址:
https://www.nature.com/articles/s42004-023-01090-2
来自牛津大学的课题组曾发表题目为「The amorphous state as a frontier in computational materials design」的研究,强调了机器学习在打破材料设计传统限制中的关键作用。展示了计算建模和人工智能的最新进展如何弥补非晶态固体原子尺度结构、微观性质和宏观功能之间此前缺失的联系。
论文地址:
https://www.nature.com/articles/s41578-024-00754-2
这一系列研究共同勾勒出一幅清晰的图景:材料科学正在进入智能化新时代,我们正处在一场由机器学习引领的材料研究转型之中,更重要的是,智能已经从新材料设计、合成,逐步蔓延至应用场景环节,势必将进一步推动新材料落地。
参考资料:
1.https://pubs.acs.org/doi/10.1021/cr300014x
一键获取 2023—2024 年 AI4S 领域高质量论文及深度解读文章 ⬇️









