Command Palette
Search for a command to run...
برنامج تعليمي لتسلسل النسخ الجيني للخلية الواحدة لعينة واحدة: مراقبة الجودة، والتجميع، والعرض الجيني (التفاضلي)
التاريخ
الحجم
2.42 GB
رابط الورقة البحثية
برنامج تعليمي حول تحليل تسلسل النسخ الجيني للخلية الواحدة
تأتي بيانات العينة المستخدمة في هذه الدراسة من ورقة بحثية نُشرت في مجلة Nature Medicine في يوليو 2024. لقاح نيوأنتيجين مخصص ودواء بيمبروليزوماب في سرطان الخلايا الكبدية المتقدم: تجربة من المرحلة الأولى والثانية بيانات تسلسل الخلايا المفردة للخلايا أحادية النواة في الدم المحيطي من المريض 6 (Yarchoan M et al. Nat Med. 2024;30:1044–1053.).
مقدمة إلى تسلسل الخلية الواحدة
إن تسلسل الخلية الفردية هو تقنية حيوية متطورة تسمح للعلماء بتحليل التعبير الجيني بشكل عميق على مستوى الخلايا الفردية. تبدأ العملية عادة بعينة أنسجة يتم تفكيكها أولاً إلى خلايا فردية لتشكيل تعليق خلية واحدة. وبعد ذلك، باستخدام تقنية الماء في الزيت، تم تغليف كل خلية على حدة للتأكد من بقائها مستقلة أثناء التحليل اللاحق.
في تسلسل الخلية الواحدة، يتم تمييز جزيء mRNA الخاص بكل خلية بمعرف جزيئي فريد، وهي عملية تسمى تسمية UMI (المعرف الجزيئي الفريد). وفي الوقت نفسه، سيتم تعيين نفس علامة الباركود لكل خلية، والتي تستخدم لتتبع مصدر كل خلية أثناء مرحلة تحليل البيانات. وبهذه الطريقة، يستطيع الباحثون قياس معلومات mRNA في كل خلية بدقة، وبالتالي فهم حالة التعبير الجيني لكل خلية.
1. وظيفة الباركود: تسجيل معلومات الخلية
2. تطبيق UMI في تسلسل الخلية الفردية: من خلال توفير معرف فريد لكل جزيء mRNA، فإنه لا يحسن دقة قياس التعبير الجيني فحسب، بل يقلل أيضًا من الأخطاء التي قد تحدث أثناء تضخيم تفاعل البوليميراز المتسلسل، وبالتالي تعزيز موثوقية وفعالية عملية التسلسل بأكملها.
العملية الأساسية
تتضمن عملية تسلسل الحمض النووي الريبوزي منقوص الأكسجين أحادي الخلية (scRNA-seq) بشكل أساسي عزل الخلية الواحدة والتقاطها، وتحلل الخلايا، والنسخ العكسي (تحويل الحمض النووي الريبوزي منقوص الأكسجين إلى cDNA)، وتضخيم cDNA وإعداد المكتبة
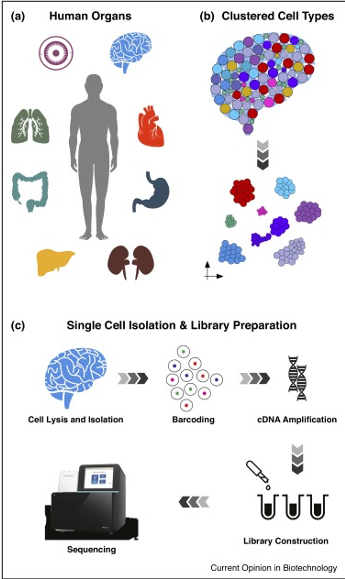
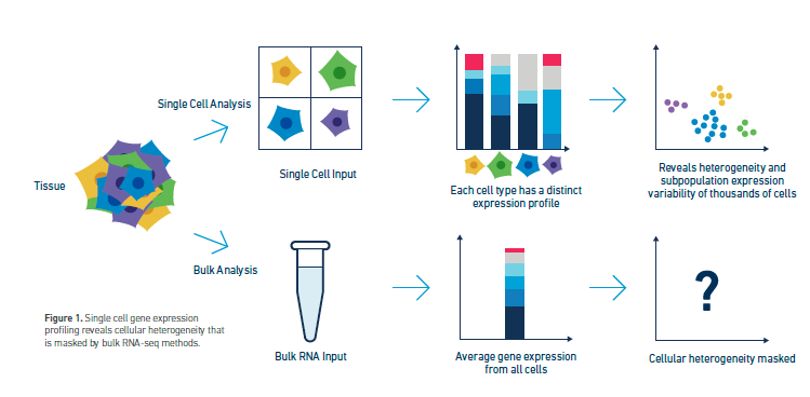
مصدر الصورة: كولكارني، أ، وآخرون. (2019). "ما وراء الحجم: مراجعة لأساليب وتطبيقات تحليل النسخ الجيني للخلايا الفردية” Curr Opin Biotechnol 58: 129-136
خطوات التشغيل
والآن سوف ننتقل إلى الجزء العملي من البرنامج التعليمي.
ابدأ تشغيل الحاوية وتحميل البيانات
1. ابدأ تشغيل الحاوية
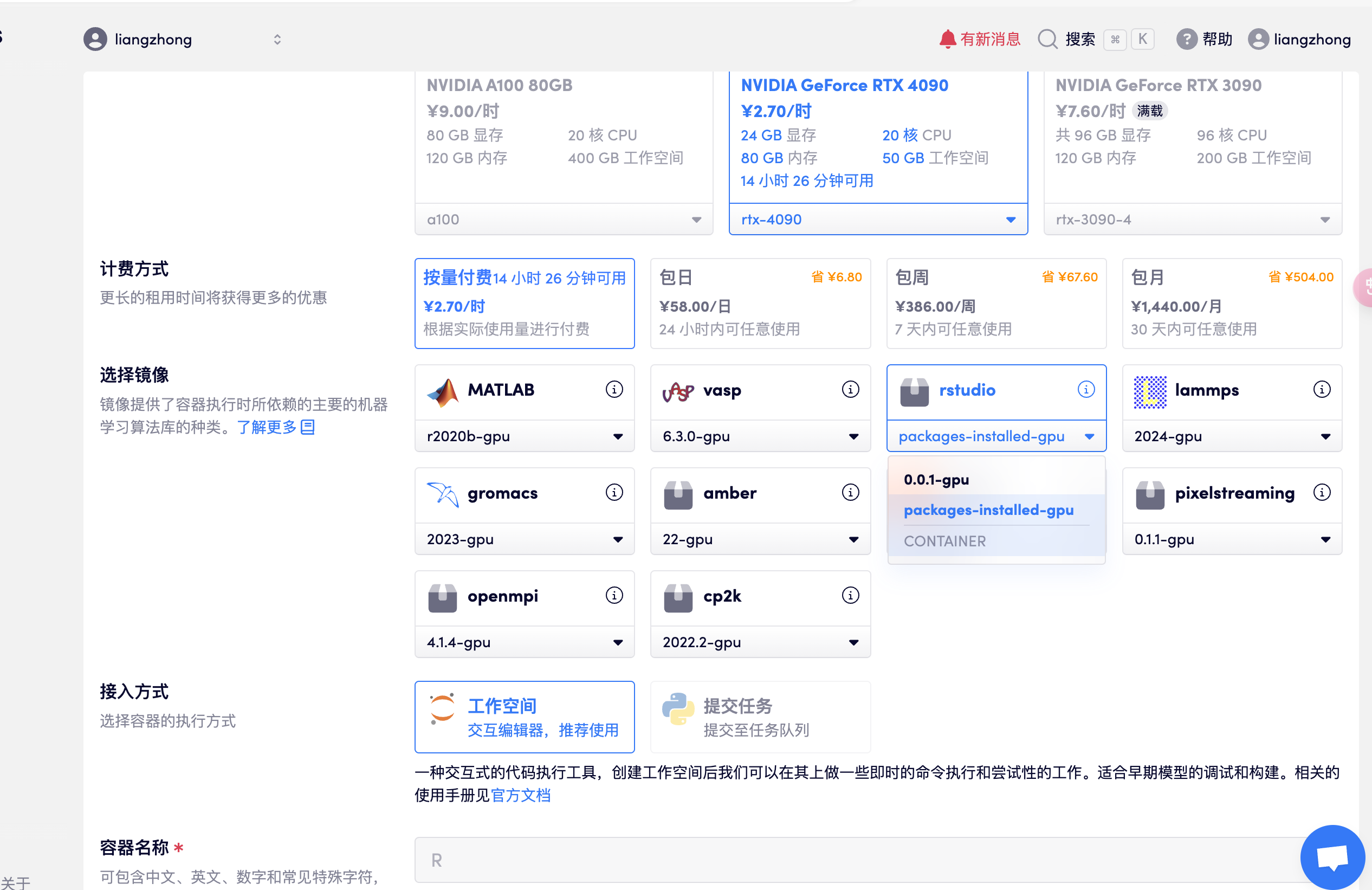
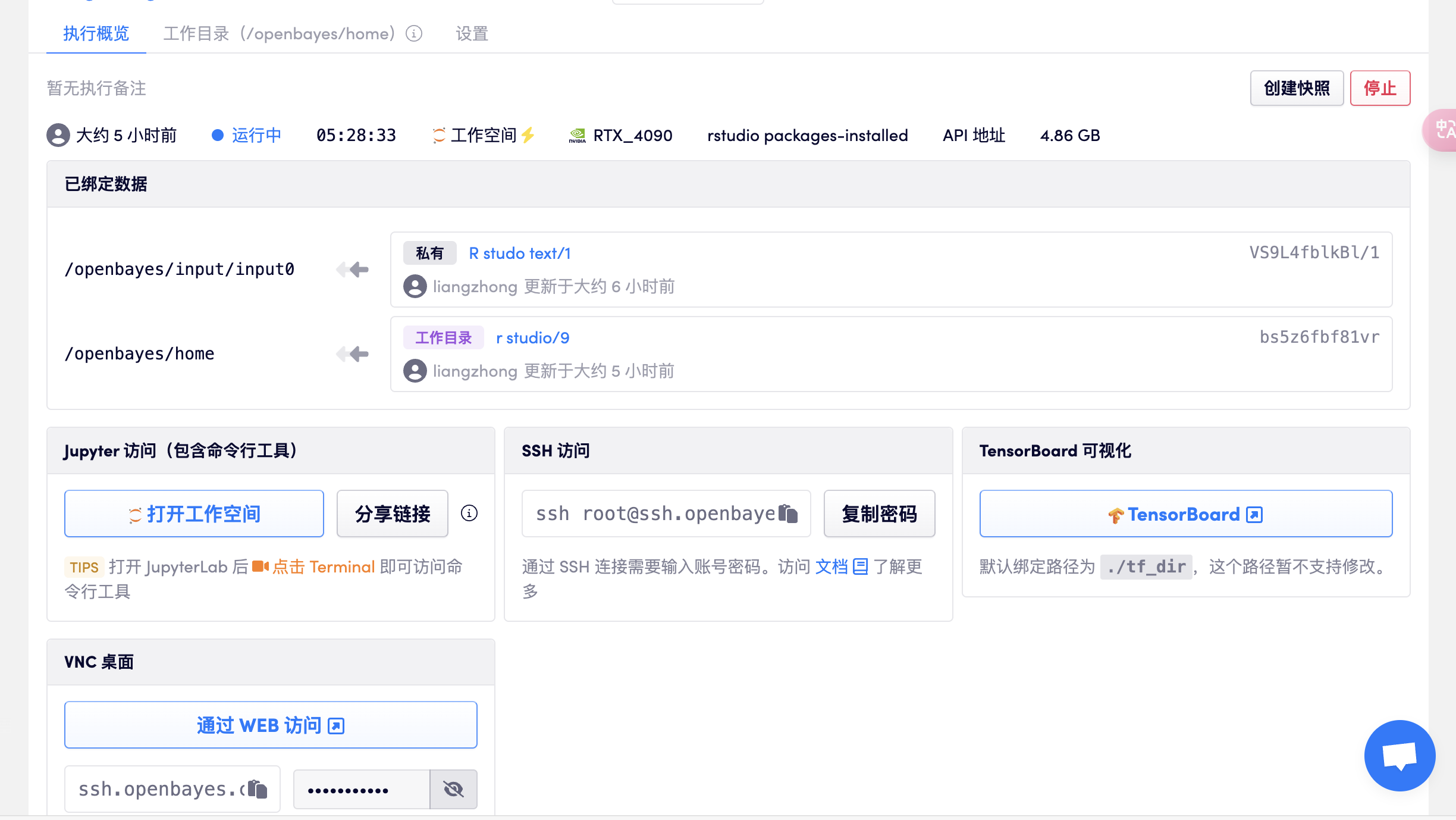
قم بإنشاء حاوية، وافتح rstudio، وحدد إصدار package-installed-gpu، وافتح عنوان API مباشرة للدخول إلى R studio بعد تخصيص الموارد (تتطلب هذه الخطوة مصادقة الاسم الحقيقي)، كما هو موضح في الشكل التالي:
- ملحوظة: بعد الدخول للبرنامج، الحساب وكلمة المرور هما: rstudio
2. تحميل البيانات
قم بتحميل البيانات المعدة مسبقًا (يمكن تنزيل البيانات واستخدامها في مجلد البيانات في دليل العمل):
(1) البيانات الخام للخلايا الفردية التي تم تسلسلها بواسطة Illumina NextSeq 500: barcodes.tsv.gz، features.tsv.gz، matrix.mtx.gz
(2) بيانات مرجعية لشرح الخلية: pbmc_multimodal.h5seurat، ref_Human_all.RData
#存放的目录如下
#此处可新建自定义文件夹,指定输入输出文件目录,本教程以 output 为例
rstudio@****(用户名)-bs5z6fbf81vrmain:~/home/output/data$ ls
barcodes.tsv.gz features.tsv.gz matrix.mtx.gz
rstudio@****(用户名)-bs5z6fbf81vr-main:~/home/output/reference_annotation$ ls
pbmc_multimodal.h5seurat ref_Human_all.RDataبعد ذلك، قم بإدخال الكود في R studio خطوة بخطوة وفقًا لخطوات البرنامج التعليمي.
#报错改为英文
Sys.setenv(LANGUAGE = "en")
options(stringsAsFactors = FALSE)
#清空所有数据
rm(list=ls())
set.seed(100)#!!!设置随机数,保证后续实验的可重复性1. إعداد كائن Seurat
# I. Setup -----------------------------------------------------------------------
# ## a. Load libraries ----------------------------------------------------------------------
```{r libraries}
library(Seurat)
library(SeuratObject)
library(sctransform)
library(patchwork)
library(Seurat)
library(SeuratData)
library(patchwork)
library(SeuratData)
library(ggplot2)
library(tidyverse)
library(glmGamPoi)
library(tools)
library(dplyr)
library(patchwork)2. تحميل البيانات
# II. Load data-----------------------------------------------------------------------
### a. Load data-----------------------------------------------------------------
getwd()#查看当前工作目录
setwd("~/home/output/results")
#修改工作目录,此处可根据自定义文件夹,指定输入输出文件目录,本教程以 output 为例
pbmc.data <- Read10X(data.dir ="~/home/output/data")#设置 10x 原始数据的工作目录,读取
seurat <- CreateSeuratObject(counts = pbmc.data, project = "pbmc", min.cells = 3, min.features = 200)#创建 seurat 对象
seurat
An object of class Seurat
16356 features across 10066 samples within 1 assay
Active assay: RNA (16356 features, 0 variable features)
1 layer present: counts
#该样本有 10066 个细胞 ,10066 个单细胞的 RNA 表达数据,测序了 16356 个基因
#counts: 通常表示原始的基因表达计数数据
colnames([email protected])
[1] "orig.ident" "nCount_RNA" "nFeature_RNA" "percent.mt" "RNA_snn_res.0.1" "seurat_clusters"
[7] "celltype"
تظهر بيانات pbmc.data في الشكل أدناه، مع عدد القراءات، وأسماء الصفوف هي الجينات، وأسماء الأعمدة هي رموز الخلايا.
pbmc.data[c("CD3D", "CD8A", "PTPRC"), 1:30] مصفوفة متفرقة 3 × 30 من فئة "dgCMatrix" [[ إخفاء أسماء الأعمدة الثلاثين 'AAACCTGAGCGAAGGG-1', 'AAACCTGAGGCCCTTG-1', 'AAACCTGAGGGTTTCT-1' … ]]
CD3D 2 . 3 6 4 1 1 3 . 6 3 4 2 1 2 1 . 2 2 5 2 3 . 6 . 4 3 CD8A . 6 . . . 6 PTPRC 1 2 2 2 3 5 3 3 3 1 3 . 2 4 1 . 1 . 2 9 . 3 4 . 1 2 . 2 1 4
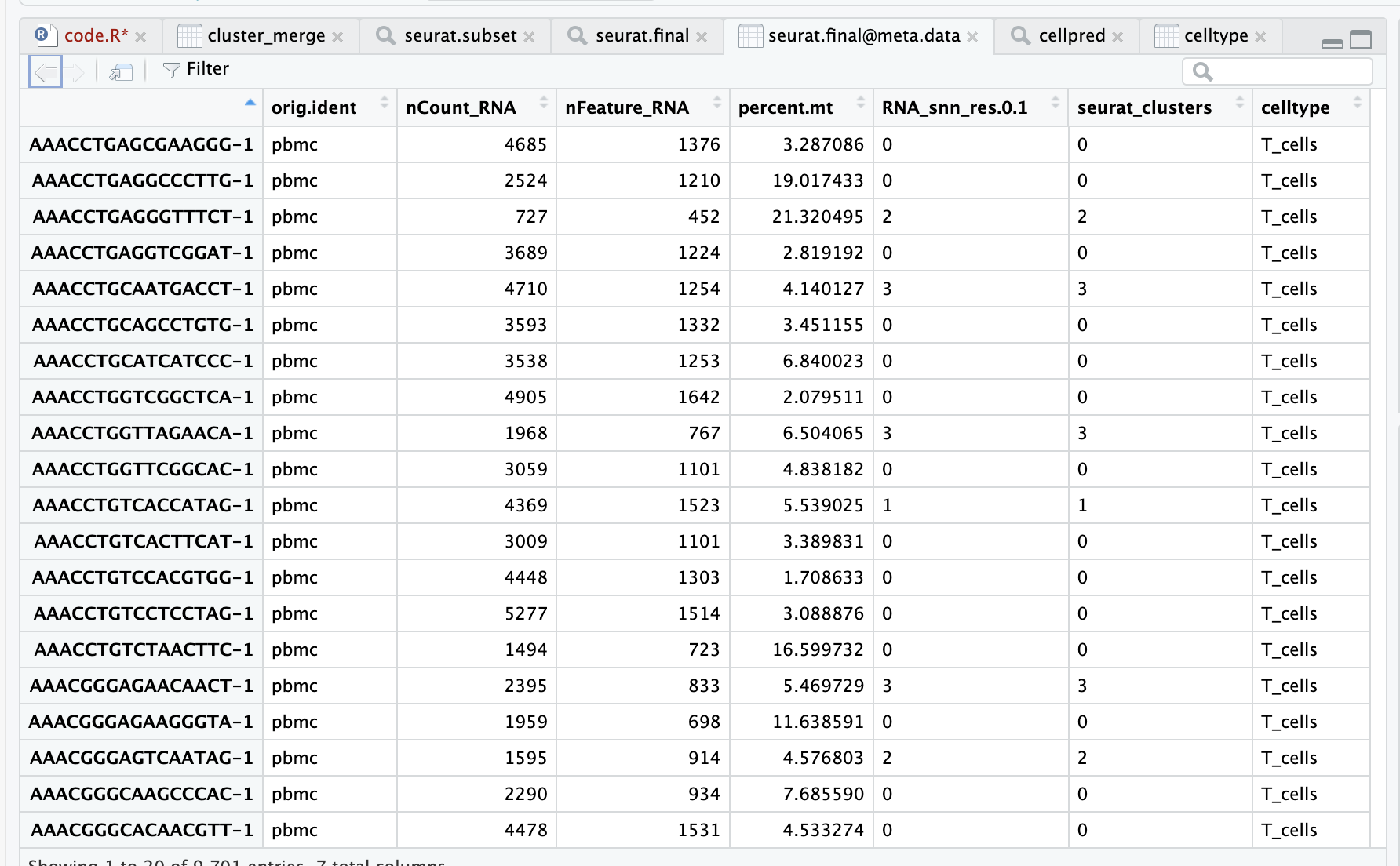
- وصف معلومات الجدول:
- اسم الصف هو الباركود
- orig.ident هي معلومات العينة،
- nCount_RNA هو إجمالي التعبير الجيني
- nFeature_RNA هو عدد الجينات
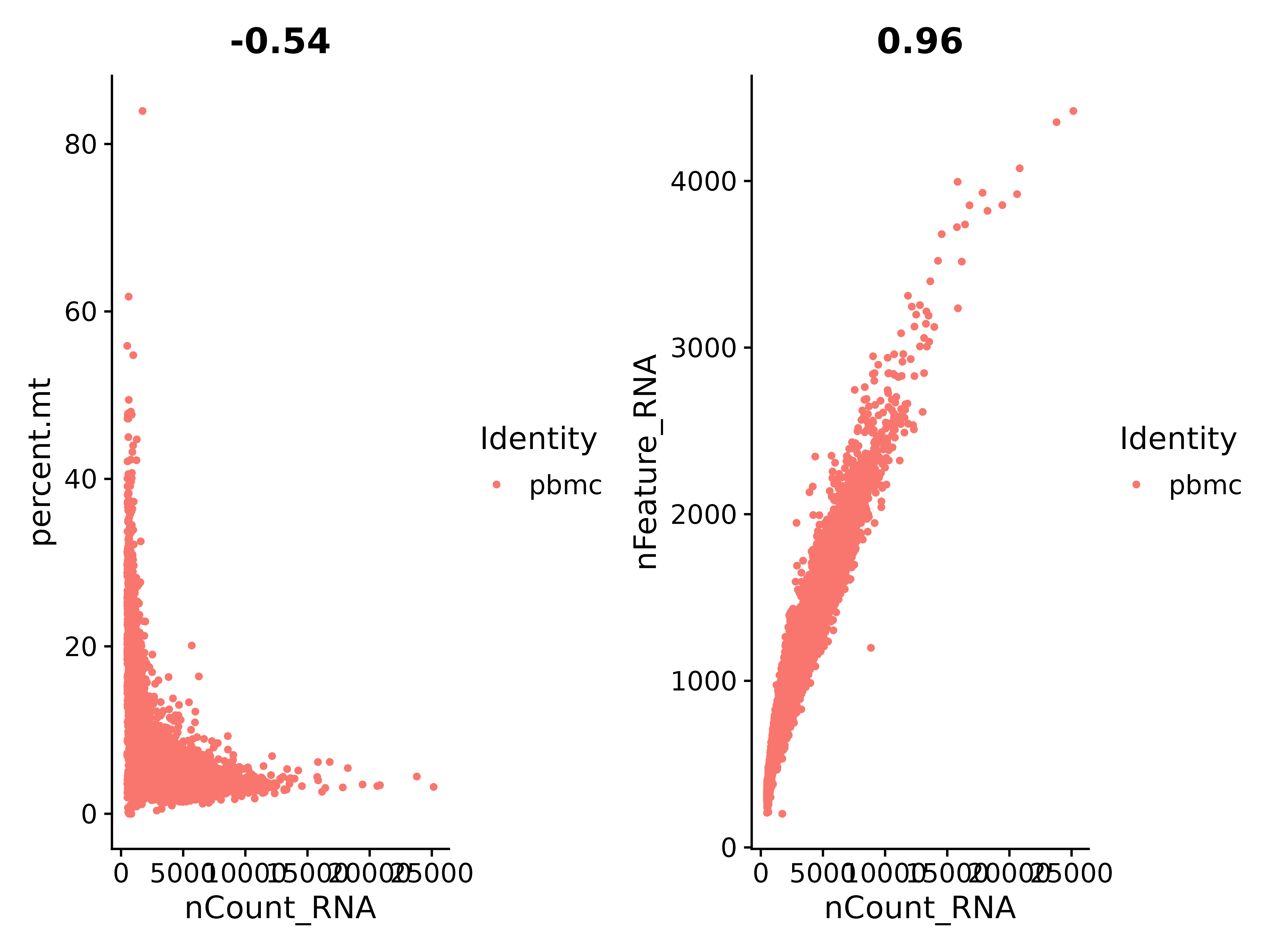
وفقًا للشكل أعلاه، دعنا نتحقق من النسبة بين nCount_RNA و percent.mt (الجين الميتوكوندريا):
يمكننا أن نرى أن nCount_RNA وnFeature_RNA متناسبان بشكل إيجابي، مع معامل 0.96، والارتباط جيد جدًا.
نصائح: جميع الجينات التي تبدأ بـ MT- هي جينات الميتوكوندريا.
3. المعالجة المسبقة القياسية
وتغطي الخطوات التالية سير عمل المعالجة المسبقة القياسية لبيانات scRNA-seq في Seurat. تتضمن هذه العمليات تحديد الخلايا وتصفيتها استنادًا إلى مقاييس مراقبة الجودة، وتطبيع البيانات وتوسيع نطاقها، واكتشاف الميزات شديدة التغير.
يمكن لـ Seurat عرض مقاييس مراقبة الجودة وتصفية الخلايا استنادًا إلى أي معايير محددة من قبل المستخدم.
# III. Pre-processing----------------------------------------------------------------
### a. Plots for percent mito, nFeature---------------------------------------------
```{r preprocessing}
# The [[ operator can add columns to object metadata. This is a great place to stash QC stats
seurat <-PercentageFeatureSet(seurat, pattern = "^MT-", col.name = "percent.mt")
# Use FeatureScatter is typically used to visualize feature-feature relationships, but can be used
# for anything calculated by the object, i.e. columns in object metadata, PC scores etc.
plot1 <- FeatureScatter(seurat, feature1 = "nCount_RNA", feature2 = "percent.mt",)
plot2 <- FeatureScatter(seurat, feature1 = "nCount_RNA", feature2 = "nFeature_RNA")
plot1 + plot2
ggsave("质控_1.png", plot1 + plot2, width = 8, height = 6, dpi=600)
#plot3 <- FeatureScatter(seurat, feature1 = "nCount_RNA", feature2 = "percent.ribo")
#plot3
بعد ذلك، نقوم بتصور مقاييس مراقبة الجودة واستخدامها لتصفية الخلايا.
انظر إلى نسبة الجينات الميتوكوندريا واستخدم مخططات الكمان لتوضيح مؤشرات مراقبة الجودة
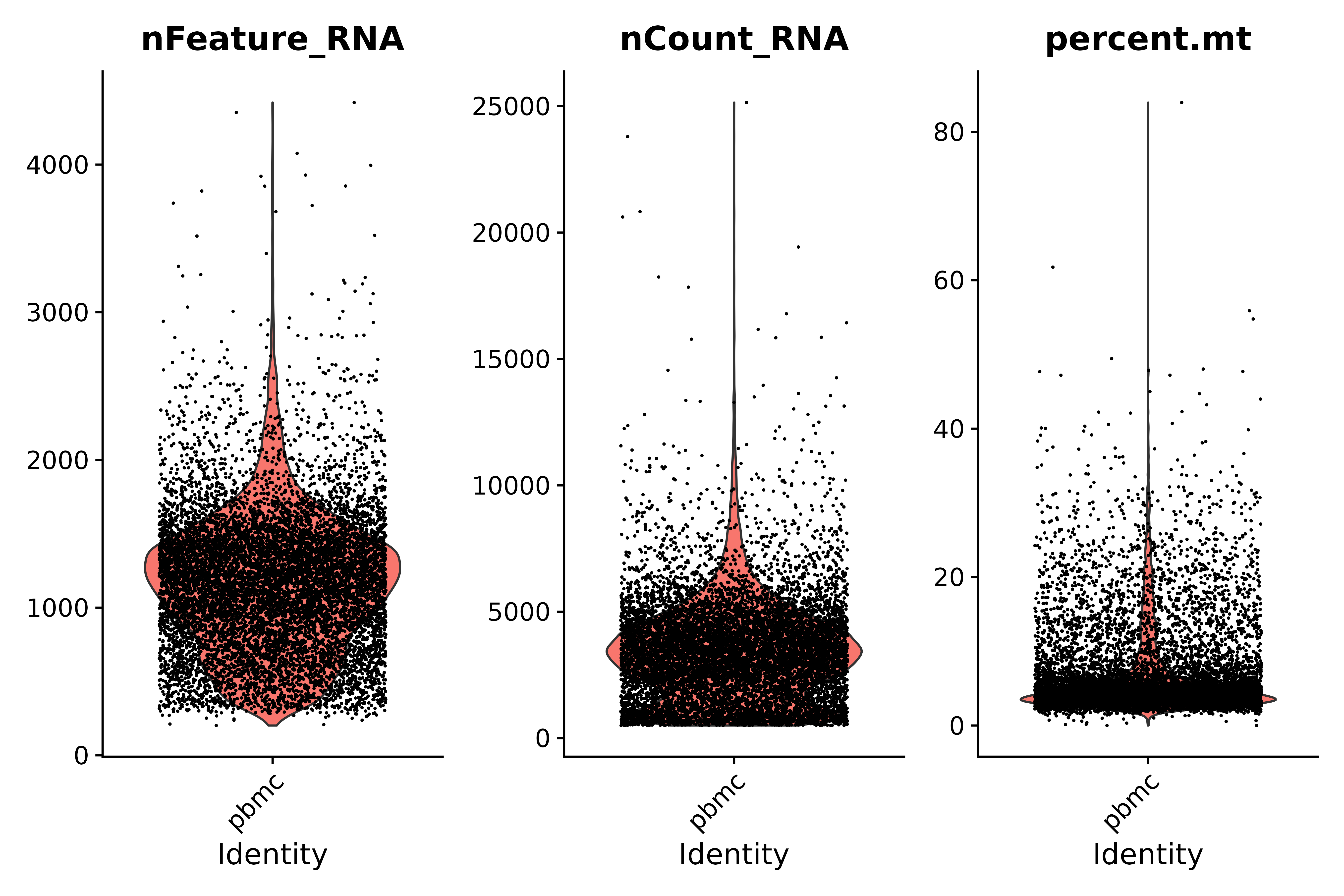
- تعليمات مراقبة الجودة:
- تحتوي الخلايا منخفضة الجودة أو القطرات الفارغة عادةً على عدد قليل جدًا من الجينات، حيث يكون nFeature_RNA > 200 و nCount_RNA > 500.
- قد تُظهر الخلايا ثنائية الصيغة الصبغية أو متعددة الصيغة الصبغية أعدادًا عالية بشكل غير طبيعي من الجينات؛ وفقًا لمخططنا، nFeature_RNA < 2500 و nCount_RNA > 500
- عادةً ما تُظهر الخلايا منخفضة الجودة/المحتضرة تضخمًا واسع النطاق في الميتوكوندريا، النسبة المئوية للكتلة < 25
# Visualize QC metrics as a violin plot
vln1 <- VlnPlot(seurat, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"#,"percent.ribo"
),ncol=3)
vln1
ggsave("质控_2.png", vln1, width = 9, height = 6, dpi=600)seurat.subset <- subset(seurat, subset = nFeature_RNA > 200 & nFeature_RNA < 2500 & nCount_RNA > 500 & nCount_RNA < 10000 &percent.mt < 25)
vln3 <- VlnPlot(seurat.subset, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)
vln3
ggsave("质控_3.png", vln3, width = 9, height = 6, dpi=600)
pdf("qc_plots.pdf");plot1 + plot2;vln1;vln3;dev.off()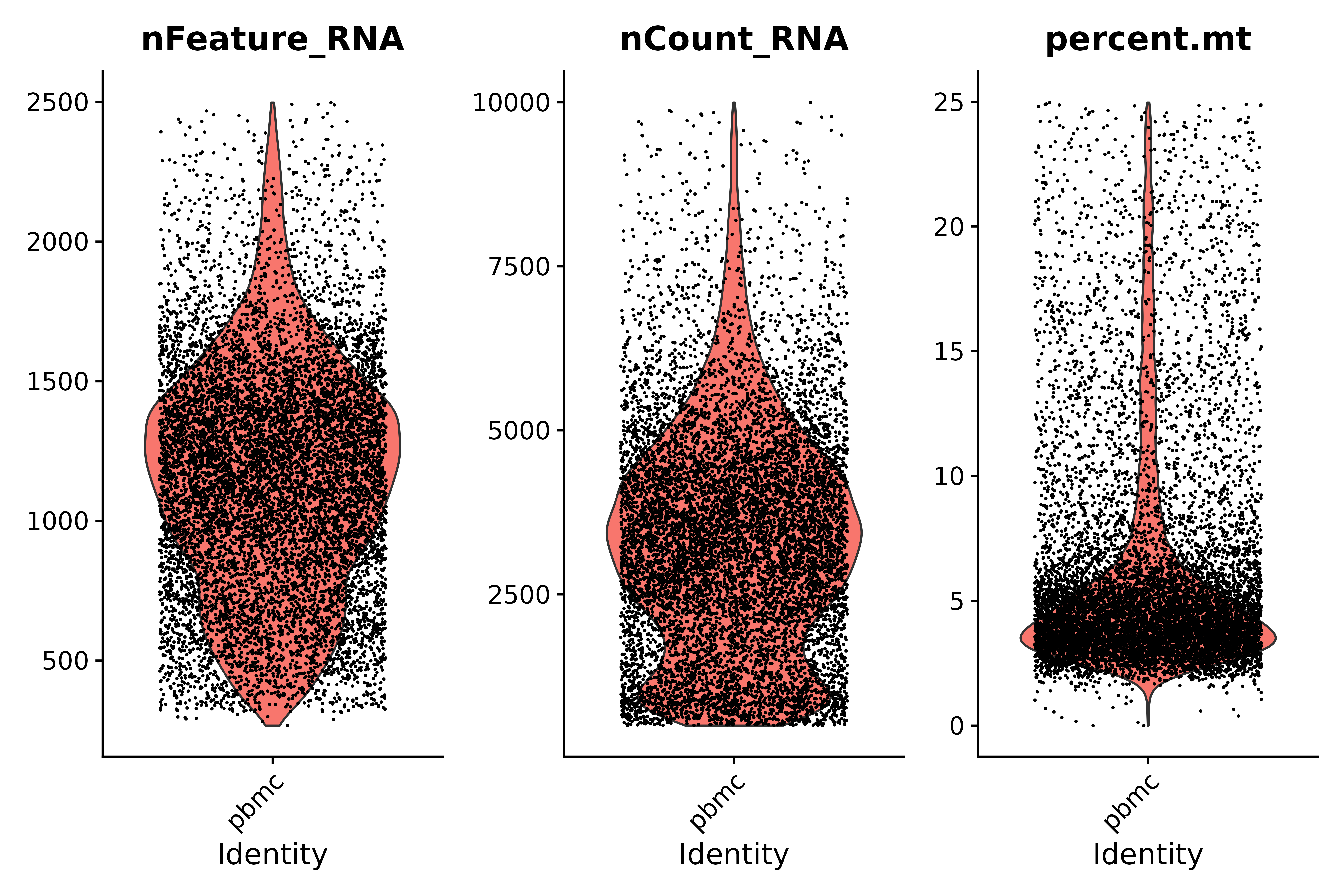
4. تطبيع البيانات وتحديد الميزات شديدة التغير (اختيار الميزة)
في تحليل بيانات النسخ الجيني للخلية الواحدة، نقوم أولاً بتطبيع البيانات للتأكد من إمكانية مقارنة مستويات التعبير الجيني بين الخلايا المختلفة بشكل فعال.
1. تطبيع البيانات
- تطبيع البيانات: نقوم أولاً بتقسيم مستوى التعبير (العدد) لجين معين في كل خلية على مستوى التعبير الإجمالي (العدد الإجمالي) للخلية. تهدف هذه الخطوة إلى إزالة الاختلاف في عمق التسلسل بين الخلايا بحيث يمكن مقارنة مستويات التعبير الجيني للخلايا المختلفة على نفس الأساس.
- ضبط المقياس: يتم بعد ذلك ضرب قيمة التعبير الطبيعية بعامل المقياس، هنا 10000، من أجل ضبط قيمة التعبير الجيني إلى مقدار أكثر ملاءمة للتحليل اللاحق.
- التحويل اللوغاريتمي: أخيرًا، أجرينا تحويلًا لوغاريتميًا على قيم التعبير المعدلة، مما ساعد على استقرار التباين بشكل أكبر وجعل الاختلافات بين الجينات ذات التعبير العالي والمنخفض أكثر وضوحًا، مما وضع الأساس لخطوات التحليل اللاحقة.
- التطبيع: من خلال الخطوات المذكورة أعلاه، قمنا بتطبيع قيم التعبير الجيني إلى مستوى كل 10000 UMIs، مما يساعد على القضاء على تأثير الاختلافات في عمق التسلسل بين الخلايا.
2. بعد ذلك، نبحث عن ميزات الطفرة:
بعد التطبيع والتوحيد القياسي، حددنا الجينات شديدة التغير من خلال تحليل تباين التعبير الجيني. تتمتع هذه الجينات بمستويات تعبير مختلفة بشكل كبير في الخلايا المختلفة وقد يتم التعبير عنها بشكل كبير في بعض الخلايا ولكن يتم التعبير عنها بمستويات أقل في خلايا أخرى.
# IV. Normalize Data----------------------------------------------------------------
seurat.subset <- NormalizeData(seurat.subset, normalization.method = "LogNormalize", scale.factor = 10000)
seurat.subset <- FindVariableFeatures(seurat.subset, selection.method = "vst", nfeatures = 2000)
# Identify the 10 most highly variable genes
top10 <- head(VariableFeatures(seurat.subset), 10)
# plot variable features with and without labels
plot3 <- VariableFeaturePlot(seurat.subset)
plot4 <- LabelPoints(plot = plot3, points = top10, repel = TRUE)
ggsave("VariableFeaturePlot_4.png", plot3 + plot4, width = 12, height = 6, dpi=600)
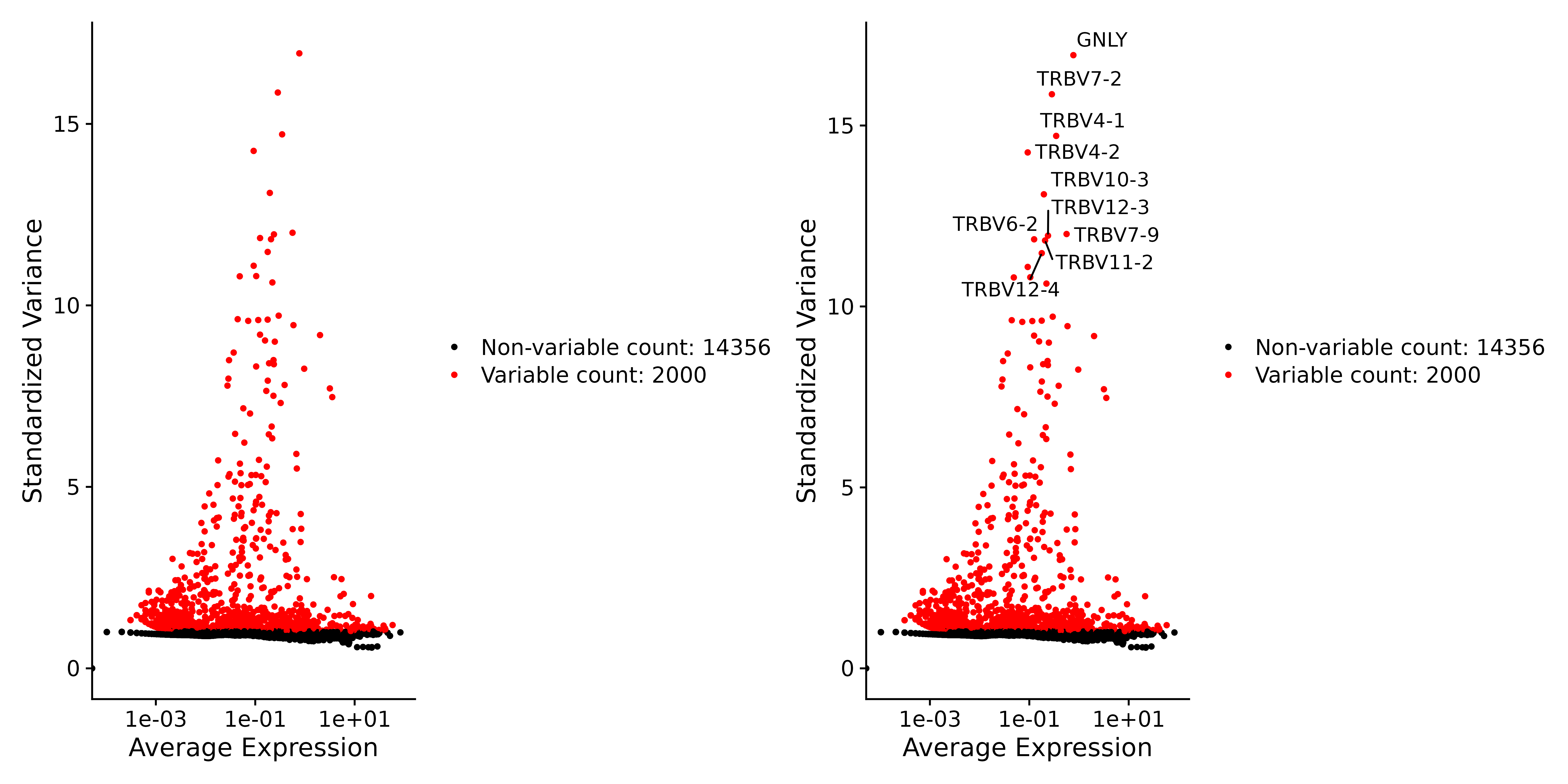
5. توسيع نطاق البيانات وتشغيل تحليل المكونات الرئيسية (PCA)
في تحليل بيانات النسخ الجيني للخلية الواحدة، يمكن لتوسيع نطاق البيانات وتحليل المكونات الرئيسية تقليل أبعاد البيانات بشكل فعال، واستخراج المصادر الرئيسية للتباين بين الخلايا، وتوفير معلومات مهمة لفهم عدم تجانس الخلايا ووظيفتها.
1. مقياس البيانات: مقياس البيانات هو تحويل خطي يهدف إلى تحويل بيانات التعبير الجيني إلى توزيع طبيعي قياسي، أي أن متوسط البيانات هو 0 والانحراف المعياري هو 1. يساعد هذا التحويل على تقليل الاختلافات بين مستويات التعبير للجينات المختلفة ويجعل البيانات أكثر تطبيعًا. من خلال التوسع، يمكن تقليل تأثير القيم المتطرفة أو القيم الشاذة في البيانات، مما يجعل توزيع البيانات أكثر توازناً، ويوفر مدخلات أكثر استقرارًا لخطوات التحليل اللاحقة. تعتبر هذه الخطوة أيضًا بمثابة تحضير لتحليل المكونات الأساسية (PCA)، لأن أداء PCA يكون أفضل على البيانات القياسية.
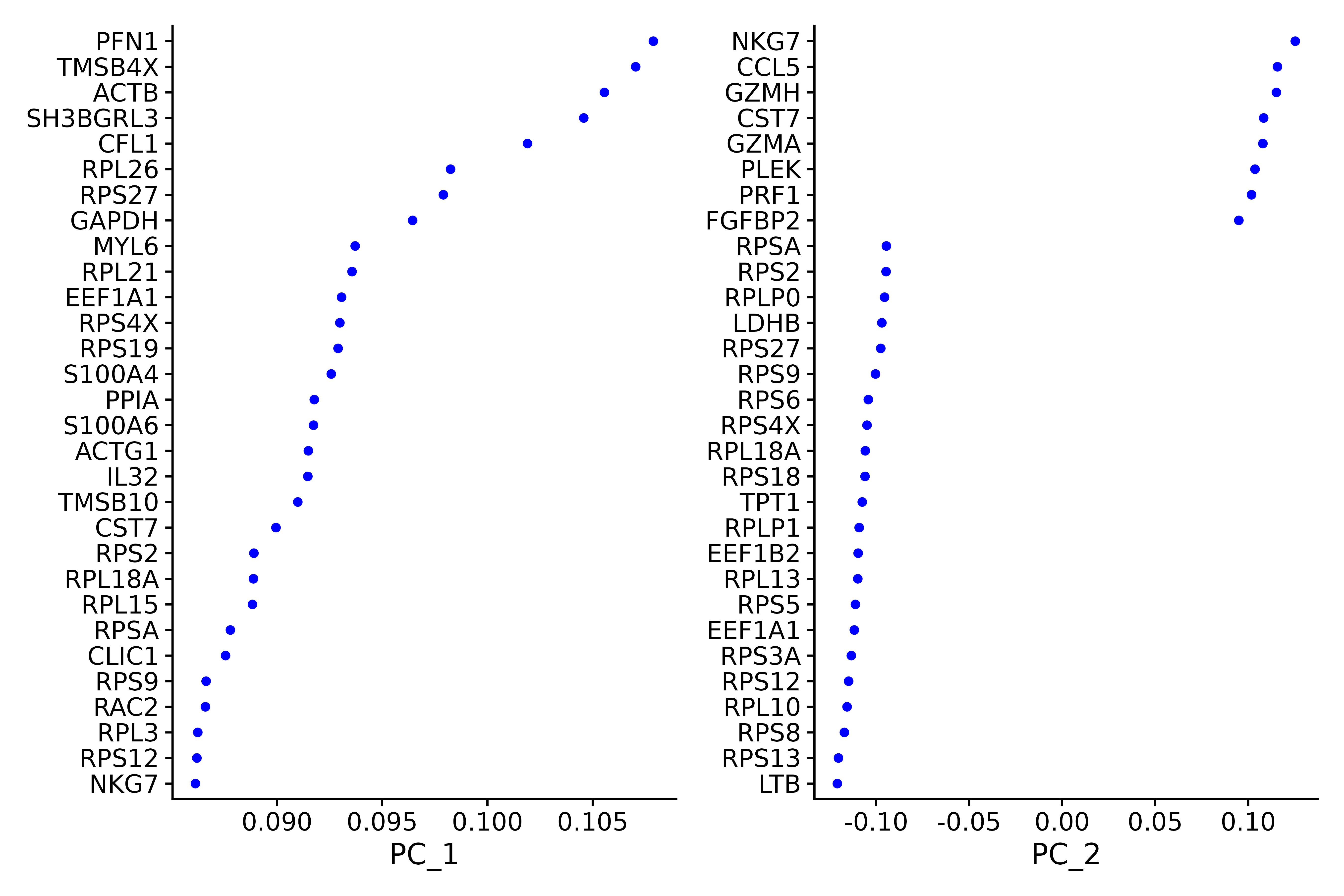
2. تشغيل PCA:
- PCA هي طريقة إحصائية تقوم بتحويل البيانات إلى نظام إحداثيات جديد عن طريق تحويل المحاور المتعامدة بحيث يكون أول أكبر تباين لأي إسقاط للبيانات على الإحداثي الأول (يسمى المكون الرئيسي الأول)، وثاني أكبر تباين يكون على الإحداثي الثاني، وهكذا.
- في تحليل النسخ أحادي الخلية، يتم استخدام PCA لفصل الخلايا الفردية على أساس الاختلافات في التعبير الجيني. يمكن النظر إلى كل خلية كنقطة في مساحة عالية الأبعاد، ويقوم تحليل المكونات الرئيسية بإعادة تمثيل هذه النقاط في مساحة منخفضة الأبعاد مع الاحتفاظ بأكبر قدر ممكن من معلومات التباين.
- يمثل كل مكون رئيسي (PCA) بشكل أساسي "سمة أساسية" للخلية، أي اتجاهًا رئيسيًا لتباين الخلية. في نتائج تحليل المكونات الرئيسية، كلما كان المكون الرئيسي أكثر تقدمًا، زاد وزنه في تباين البيانات، مما يعني أنه يحتوي على مزيد من المعلومات.
- من خلال تحليل المكونات الرئيسية (PCA)، يمكن ضغط معلومات التعبير عالية الأبعاد للخلايا إلى عدة مكونات رئيسية رئيسية، والتي يمكن استخدامها لتحليل التجميع اللاحق أو التصور أو التحليل اللاحق الآخر.
all.genes <- rownames(seurat.subset)
seurat.subset <- ScaleData(seurat.subset, features = all.genes)
seurat.subset <- RunPCA(seurat.subset, features = VariableFeatures(object = seurat))##### Examine and visualize PCA results a few different ways-------------
print(seurat.subset[["pca"]], dims = 1:5, nfeatures = 5)
p1 <- VizDimLoadings(seurat.subset, dims = 1:2, reduction = "pca")
p1
ggsave("VizDimLoadings_5.png", p1, width = 9, height = 6, dpi=600)
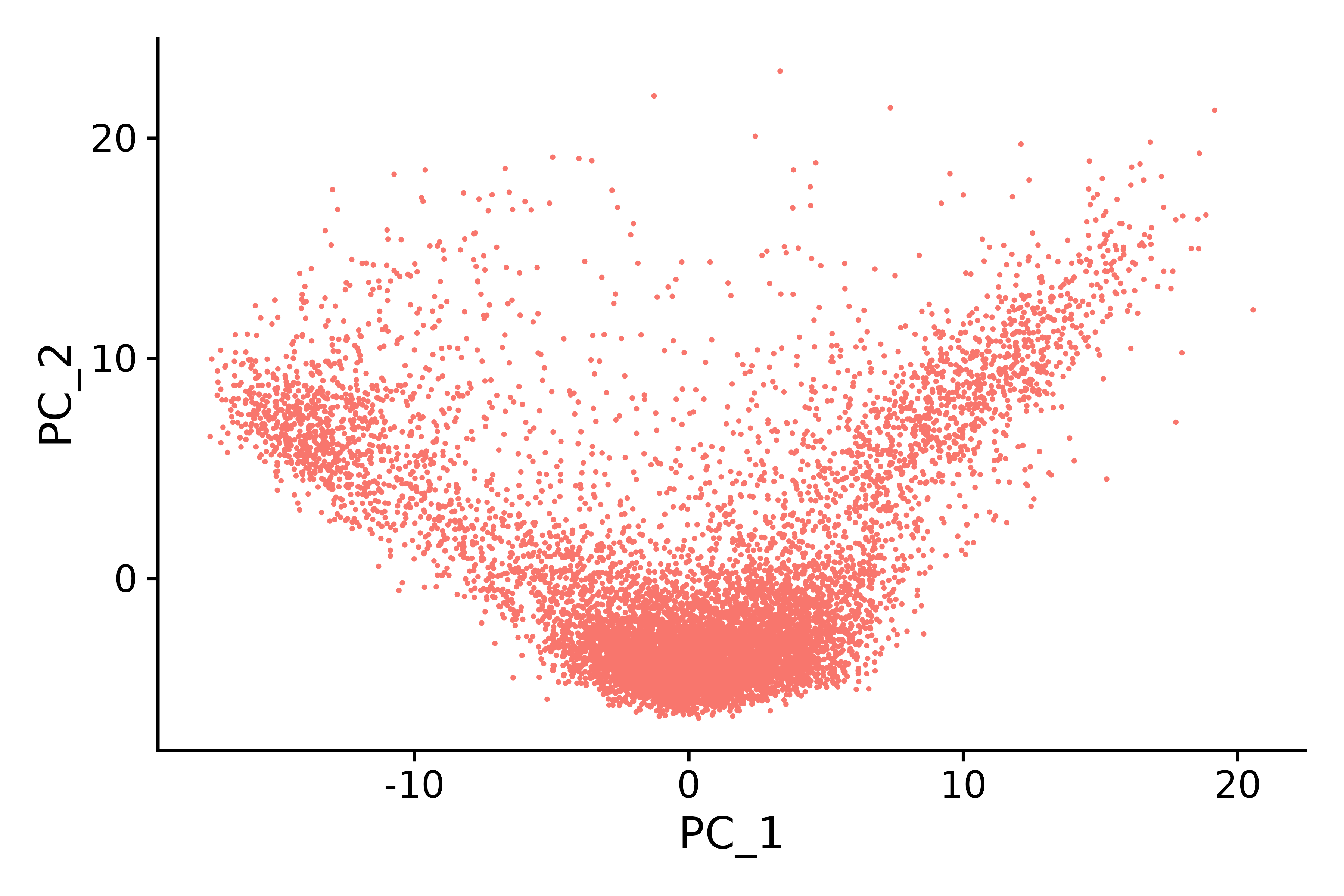
p2 <- DimPlot(seurat.subset, reduction = "pca") + NoLegend()
p2
ggsave("pca_6.png", p2, width = 6, height = 4, dpi=600)
p3 <- DimHeatmap(seurat.subset, dims = 1, cells = 500, balanced = TRUE)
p3
ggsave("pca_1dims.png", p3, width = 6, height = 4, dpi=600)
p4 <- DimHeatmap(seurat.subset, dims = 1:15, cells = 500, balanced = TRUE)
p4
ggsave("pca_15dims.png", p4, width = 10, height = 10, dpi=600)
p5 <- ElbowPlot(seurat.subset)
p5
ggsave("ElbowPlot.png", p5, width = 10, height = 10, dpi=600)
DimHeatmap() هي أداة قوية لتحليل البيانات تتيح للباحثين التعرف بسهولة على المصادر الرئيسية للتباين في مجموعة البيانات واستكشافها. بعد إجراء تحليل المكونات الأساسية (PCA)، تكون DimHeatmap() مفيدة بشكل خاص في تحديد المكونات الأساسية (PCS) التي يجب تضمينها لإعلام التحليل المتعمق اللاحق. تتضمن هذه العملية تصنيف الخلايا والميزات وفقًا لدرجات تحليل المكونات الرئيسية (PCA)، مما يضمن دقة وأهمية التحليل.
بالإضافة إلى ذلك، تسمح DimHeatmap() للمستخدم بتعيين عدد محدد من الخلايا لرسم خلايا "متطرفة" على وجه التحديد تعرض خصائص متطرفة في كلا طرفي طيف البيانات. يعمل هذا النهج على تحسين كفاءة التخطيط بشكل كبير عند العمل مع مجموعات بيانات كبيرة لأنه يركز على الأجزاء الأكثر أهمية من البيانات، مما يؤدي إلى تسريع عملية التصور بأكملها.
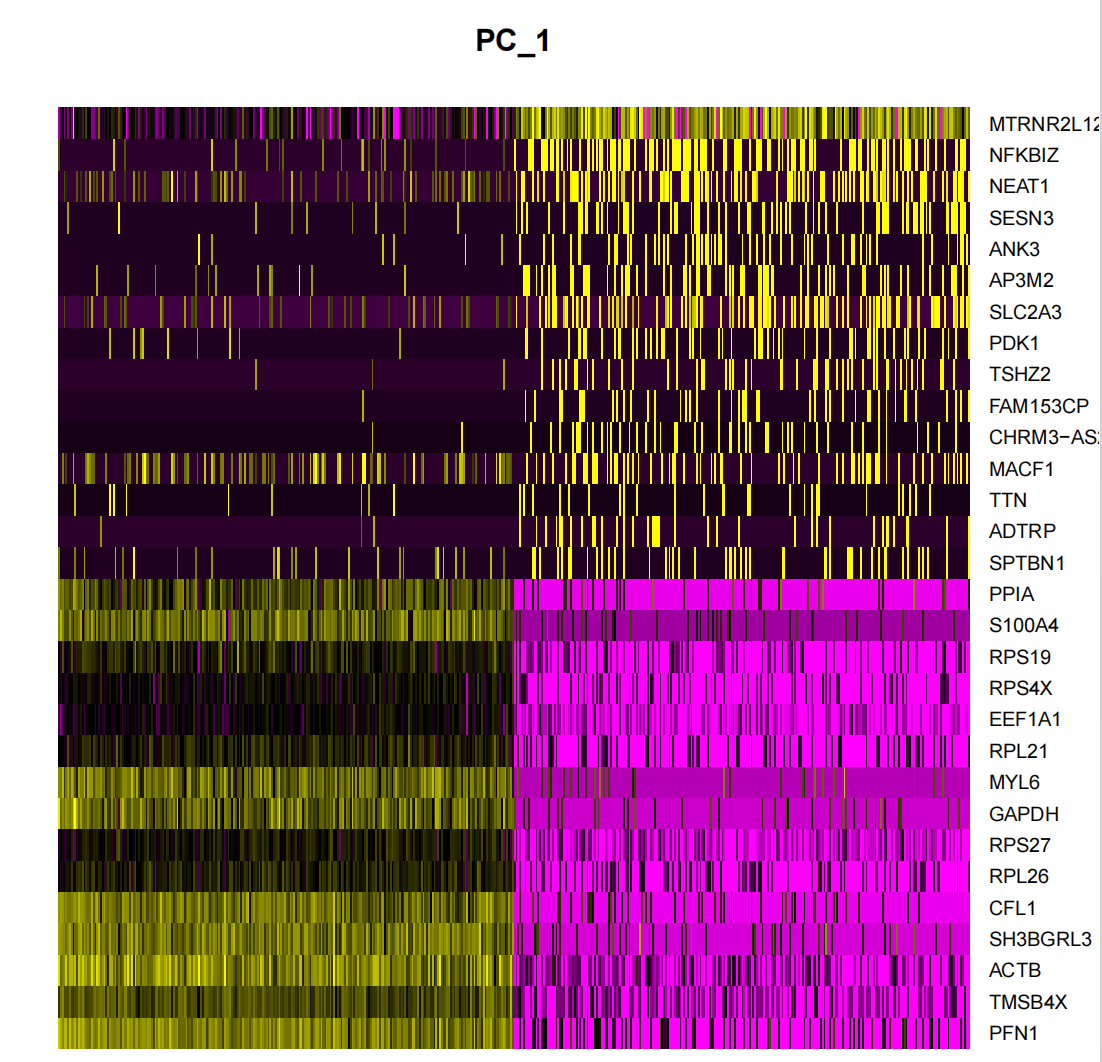

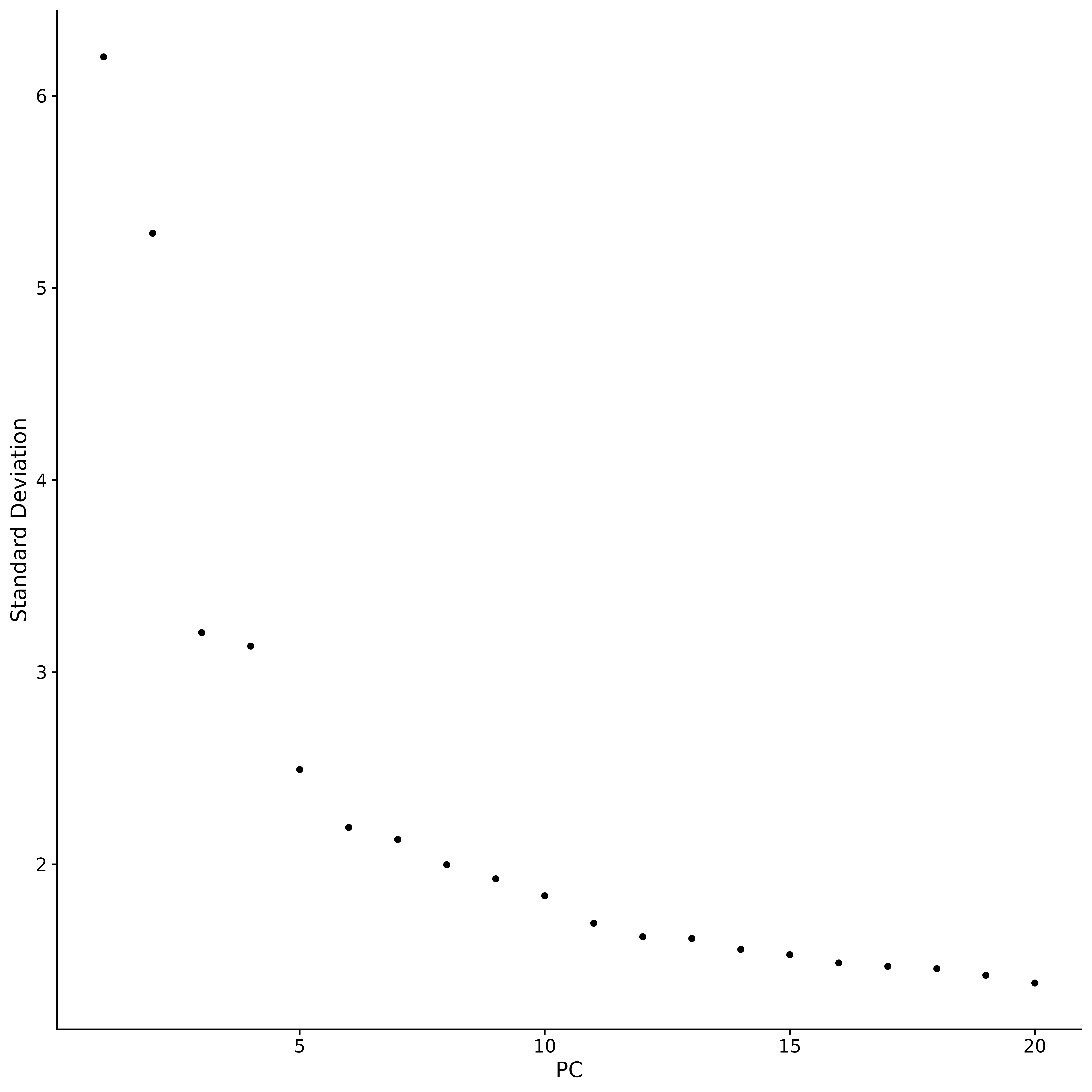
من الممارسات الشائعة في تحليل المكونات الأساسية (PCA) استخدام "مخطط الكوع" لتحديد عدد المكونات الأساسية التي يجب الاحتفاظ بها للتحليل اللاحق. يوضح الرسم البياني أن معظم الإشارة الحقيقية يتم التقاطها في أول 17 مكونًا رئيسيًا. في مخطط الكوع، نبحث عن نقطة التحول، "الكوع". قبل هذه النقطة، يعمل كل مكون رئيسي إضافي على تحسين القدرة التفسيرية للبيانات بشكل كبير، ولكن بعد هذه النقطة، تبدأ المكاسب من إضافة المكونات الرئيسية في التضاؤل.وهذا يشير إلى أنه تم الوصول إلى حالة التوازن.. وبناءً على نتائج تحليل مخطط الكوع، قمنا باختيار أول 17 مكونًا رئيسيًا للتحليل اللاحق. لأن إضافة المزيد من المكونات الأساسية لن يؤدي إلى تحسين التمييز بين البيانات بشكل كبير، بل سيؤدي إلى زيادة غير ضرورية في كمية الحساب. إذا واصلنا زيادة عدد المكونات الرئيسية، فقد يتحسن التمييز بين البيانات قليلاً، لكن هذا التحسن ليس كبيراً وسوف يؤدي إلى زيادة كبيرة في التكاليف الحسابية. لذلك، فإن اختيار 17 مكونًا رئيسيًا يعد خيارًا معقولًا يوازن بين التمييز والكفاءة الحسابية.
pdf("dimreduction_plots.pdf")#保存为 pdf 文件
VizDimLoadings(seurat.subset, dims = 1:2, reduction = "pca")
DimPlot(seurat.subset, reduction = "pca")
DimHeatmap(seurat.subset, dims = 1, cells = 500, balanced = TRUE)
DimHeatmap(seurat.subset, dims = 1:15, cells = 500, balanced = TRUE)
ElbowPlot(seurat.subset)
dev.off()seurat.subset.dimReduction <- seurat.subset
seurat.subset.dimReduction <- FindNeighbors(seurat.subset.dimReduction, reduction="pca", dims = 1:17)
saveRDS(seurat.subset.dimReduction, file = "~/home/output/data/seurat_pre_FindClusters.rds")
# seurat.subset.dimReduction <- readRDS("~/home/output/data/seurat_pre_FindClusters.rds")6. تجميع الخلايا
### 3. Find clusters,聚类
```{r find_clusters}
seurat.final <- FindClusters(object = seurat.subset.dimReduction, resolution = 0.1)
head(Idents(seurat.final), 5)
seurat.final <- RunUMAP(seurat.final, reduction="pca", dims=1:17)
#Visualize with UMAP
p1<- DimPlot(seurat.final, reduction = "umap")
p1
ggsave("find_clusters.png", p1, width = 6, height =5 , dpi=600)
saveRDS(seurat.final, file = "~/home/output/data/seurat_final.rds")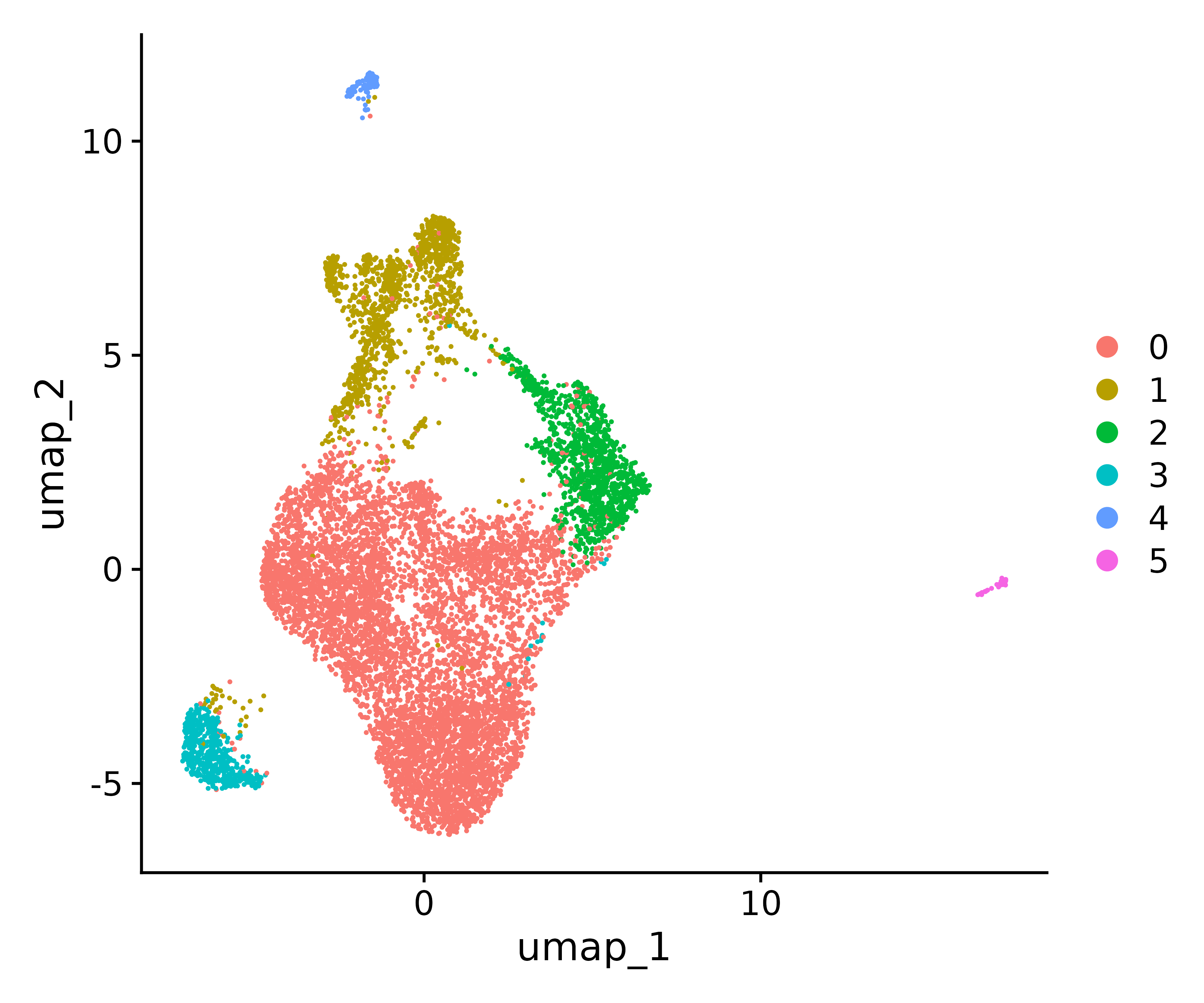
التصور
هناك ثلاثة مخططات مشتركة لعرض التعبير الجيني في مجموعات مختلفة:
1. خريطة حرارية 2. خريطة فقاعية 3. مخطط مميز
table(Idents(seurat.final))
# find markers between cluster1 and cluster 2
##用来找 cluster 之间的寻找差异表达特征
cluster2.markers <- FindMarkers(seurat.final, ident.1 = 1,ident.2 = 2)
head(cluster2.markers, n = 5)
# find markers for every cluster compared to all remaining cells, report only the positive ones
#找每个 cluster 的 marker 基因
pbmc.markers <- FindAllMarkers(seurat.final, only.pos = TRUE)
ClusterMarker_noRibo <- pbmc.markers[!grepl("^RP[SL]", pbmc.markers$gene, ignore.case = F),]
ClusterMarker_noRibo_noMito <- ClusterMarker_noRibo[!grepl("^MT-", ClusterMarker_noRibo$gene, ignore.case = F),]
###去除核糖体基因和线粒体基因的影响
ClusterMarker_noRibo_noMito %>%
group_by(cluster) %>%
dplyr::filter(avg_log2FC > 1) %>%
slice_head(n = 5) %>%
ungroup() -> top5
###展示每个 cluster 的 top5 的基因
heatmap <- DoHeatmap(seurat.final, features = top5$gene) + NoLegend()
ggsave("heatmap.png", heatmap, width = 10, height =5 , dpi=600) هذا الشكل عبارة عن خريطة حرارية توضح الجينات المميزة
هذا الشكل عبارة عن خريطة حرارية توضح الجينات المميزة
g =unique(top5$gene)
##定义 top5 的 gene 列,作为基因集 g,也可以自定义
p <- DotPlot(seurat.final,features=g)+
coord_flip() + # 翻转坐标轴
theme(
panel.grid = element_blank(), # 移除网格线
axis.text.x = element_text(angle = 0, hjust = 0.5, vjust = 0.5) # 旋转 x 轴标签
)
ggsave("allmarker_dotpot.png", p, width = 8, height =7 , dpi=600)
#气泡图展示 marker 基因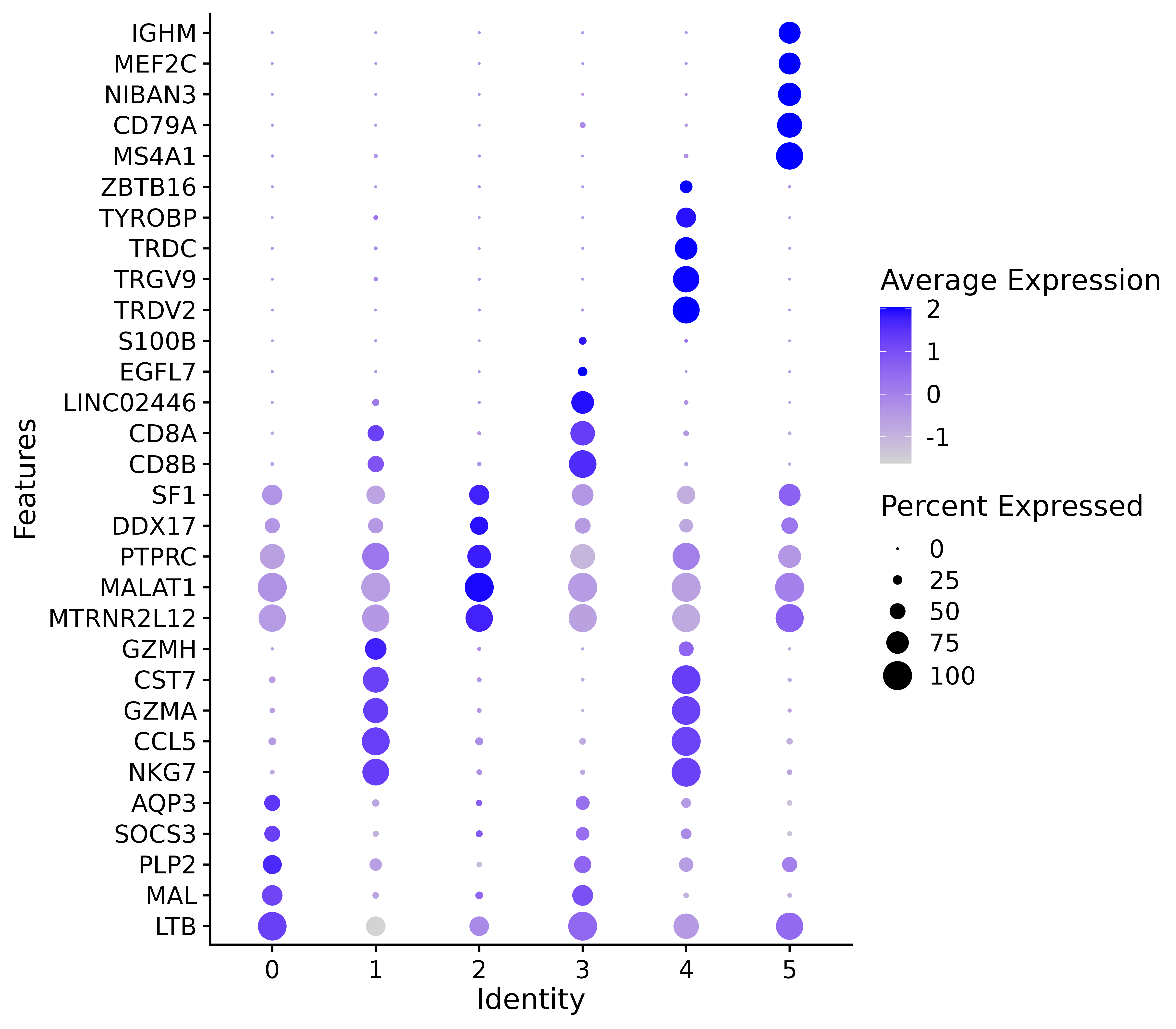
هذا الشكل عبارة عن مخطط فقاعي يوضح الجينات المميزة
# 加载 dplyr 包
library(dplyr)
# 提取 cluster 和 gene 列
cluster_gene_mapping <- top5 %>%
select(cluster, gene)
# 查看结果
head(cluster_gene_mapping,30)
write.csv(cluster_gene_mapping,"cluster_allmarkers_TOP5.csv", row.names = p <- FeaturePlot(seurat.final, features = g)
ggsave("allmarker_featurepot.png", p, width = 15, height =15 , dpi=600)
###查看自定义感兴趣的基因
p2 <- FeaturePlot(seurat.final, features = c("MS4A1", "GNLY", "CD3E", "CD14", "FCER1A", "FCGR3A", "LYZ", "PPBP",
"CD8A"))
ggsave("allmarker_featurepot2.png", p2, width = 15, height =15 , dpi=600)


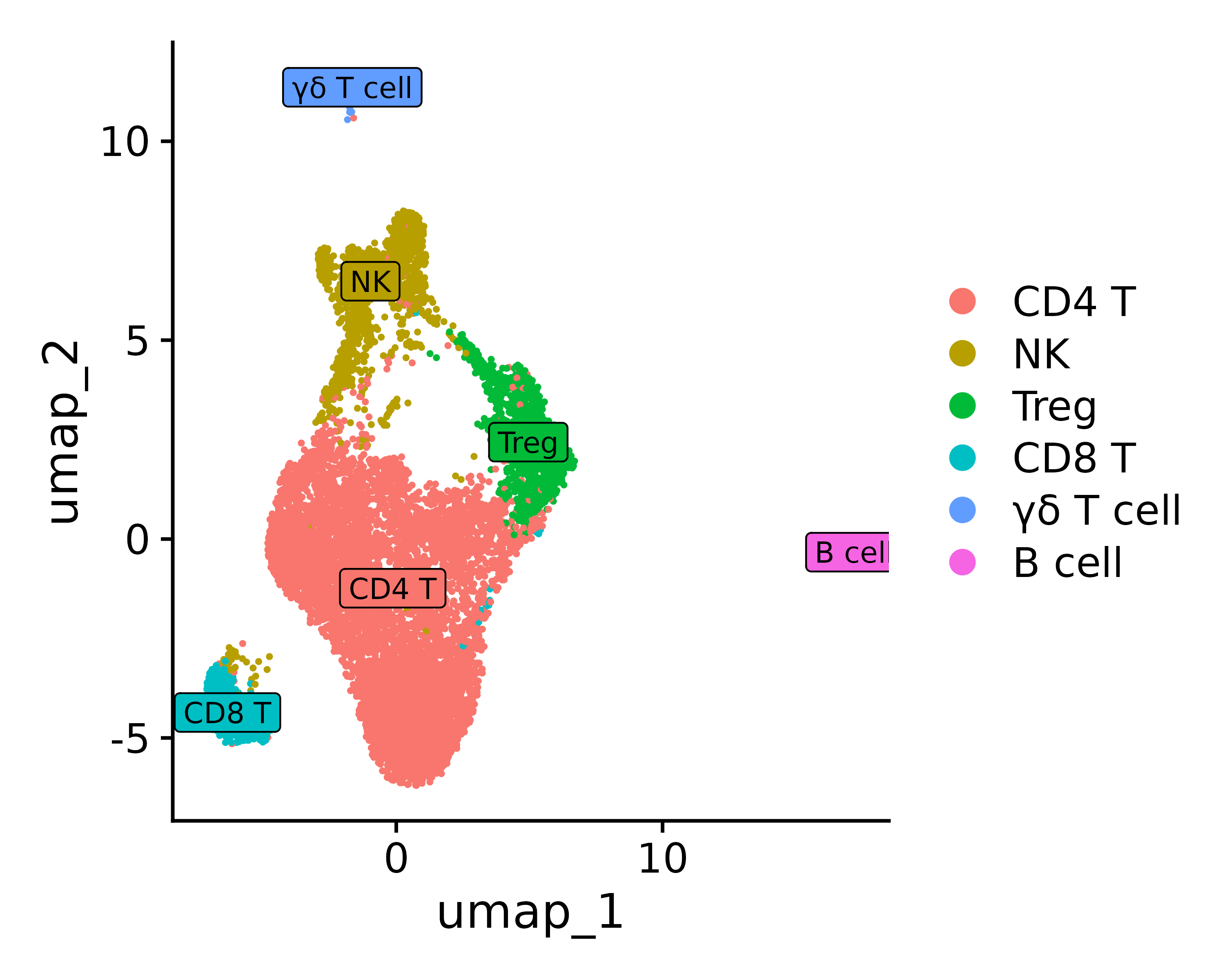
7. تحليل البيانات
توضح هذه الخطوة كيفية استخدام جينات محددة لتحديد وتعريف أنواع الخلايا في تحليل بيانات النسخ للخلية الفردية.
بافتراض أنك قمت بتحديد نوع الخلية بناءً على جينات العلامة العليا وما إلى ذلك، يمكنك تحديد نوع الهوية
0 1 2 3 4 5 "CD4 T"، "NK"، "Treg"، "CD8 T"، "خلية T γδ"، "خلية B"
(التعليقات التالية هي للإشارة فقط وليست تعليقات فعلية.)
new.cluster.ids <- c("CD4 T", "NK", "Treg", "CD8 T", "γδ T cell",
"B cell")
names(new.cluster.ids) <- levels(seurat.final)
seurat.final <- RenameIdents(seurat.final, new.cluster.ids)
p <- DimPlot(seurat.final, reduction = "umap", label = TRUE, pt.size = 0.5) + NoLegend()
p3 = DimPlot(seurat.final, reduction = "umap", label = TRUE, label.size = 3, pt.size = .3, label.box = T)
ggsave("annotation_umap.png", p, width = 5, height =4 , dpi=600)
ggsave("annotation_umap2.png", p3, width = 5, height =4 , dpi=600)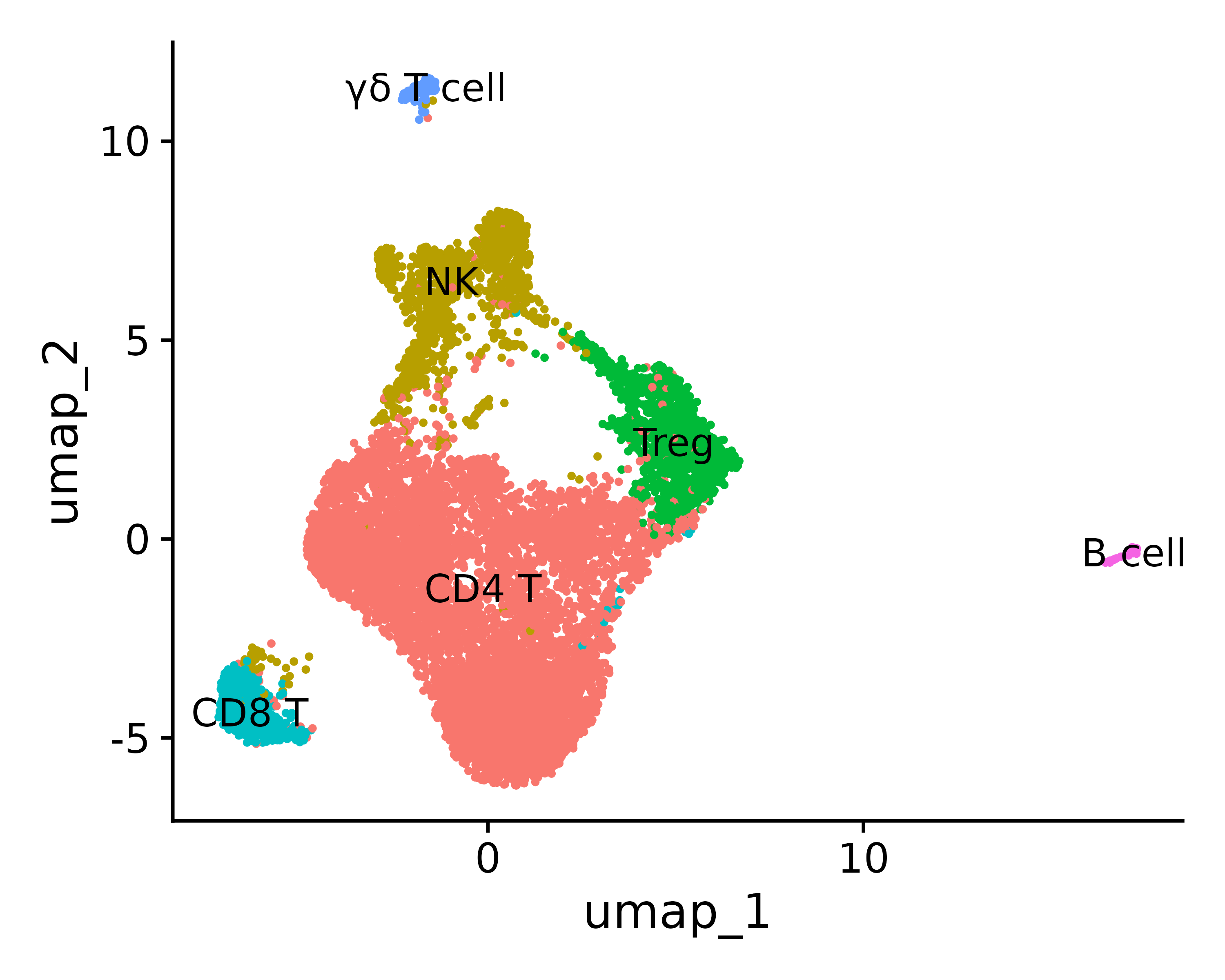
مرجع
بناء الذكاء الاصطناعي بالذكاء الاصطناعي
من الفكرة إلى الإطلاق — سرّع تطوير الذكاء الاصطناعي الخاص بك مع المساعدة البرمجية المجانية بالذكاء الاصطناعي، وبيئة جاهزة للاستخدام، وأفضل أسعار لوحدات معالجة الرسومات.