Command Palette
Search for a command to run...
برنامج GROMACS التعليمي للبدء: الليزوزيم في الماء
محاكاة الديناميكية الجزيئية باستخدام "الليزوزيم في الماء" كمثال
مقدمة البرنامج التعليمي
يعد هذا البرنامج التعليمي بمثابة برنامج تعليمي تمهيدي حول محاكاة الديناميكيات الجزيئية باستخدام برنامج GROMACS. الليزوزيم في الماء على سبيل المثال، تعلم كيفية إعداد وتشغيل محاكاة ديناميكية جزيئية لبروتين نموذجي في الماء.
لقد نجحت نسخة GPU من GROMACS التي تستخدم بطاقة الرسوميات NVIDIA RTX 4090 على منصة OpenBayes في تحسين كفاءتها الحسابية بشكل كبير بعد الحوسبة المتوازية باستخدام GPU، بسرعة تصل إلى 255ns/يوم! وهنا أداء السرعة:
Core t (s) Wall t (s) (%)
Time: 3972.923 198.659 1999.9
(ns/day) (hour/ns)
Performance: 255.471 0.094
مقدمة إلى GROMACS
GROMACS (آلة جرونينجن للمحاكاة الكيميائية) عبارة عن مجموعة برامج عالية الأداء لمحاكاة الديناميكيات الجزيئية. يتم استخدامه بشكل أساسي لنمذجة ومحاكاة سلوك حركة الجزيئات البيولوجية (مثل البروتينات والدهون والأحماض النووية) في ظل ظروف مختلفة. تم تطويره في الأصل في جامعة جرونينجن في هولندا وأصبح أحد أكثر أدوات المصدر المفتوح استخدامًا في مجال الديناميكيات الجزيئية.
الميزات الرئيسية لـ GROMACS
1. أداء عالي:
• تم تحسين GROMACS بشكل كبير للحوسبة المتوازية ويمكن تشغيله بكفاءة على أنظمة وحدة المعالجة المركزية ووحدة معالجة الرسومات متعددة النواة الحديثة.
• يدعم OpenMP وMPI للحوسبة المتعددة الخيوط والموزعة.
2. مجموعة واسعة من التطبيقات:
• يمكن استخدامها لمحاكاة سلوك الجزيئات الصغيرة والمجمعات البروتينية الكبيرة.
• يدعم الأبحاث المتعلقة بالجزيئات الحيوية والبوليمرات والمركبات غير العضوية وغيرها من الأنظمة الكيميائية.
3. المرونة:
• يوفر مجموعة غنية من الأدوات للمعالجة المسبقة (على سبيل المثال إنشاء الطوبولوجيا، والإذابة) والمعالجة اللاحقة (على سبيل المثال تحليل المسار، وحساب الطاقة).
• دعم مجالات القوة المختلفة، مثل AMBER وCHARMM وGROMOS.
4. سهولة الاستخدام:
• يتضمن GROMACS واجهة سطر أوامر سهلة الاستخدام.
• يوفر وثائق ودروس تعليمية مفصلة مناسبة للمبتدئين والمستخدمين المتقدمين.
5. مفتوح المصدر وقابل للتطوير:
• يسمح ترخيص المصدر المفتوح للمستخدمين بتعديل GROMACS وتوسيعه لتناسب احتياجات محددة.
• لديه مجتمع مستخدمين نشط وفريق تطوير.
عملية الاستخدام العامة لـ GROMACS
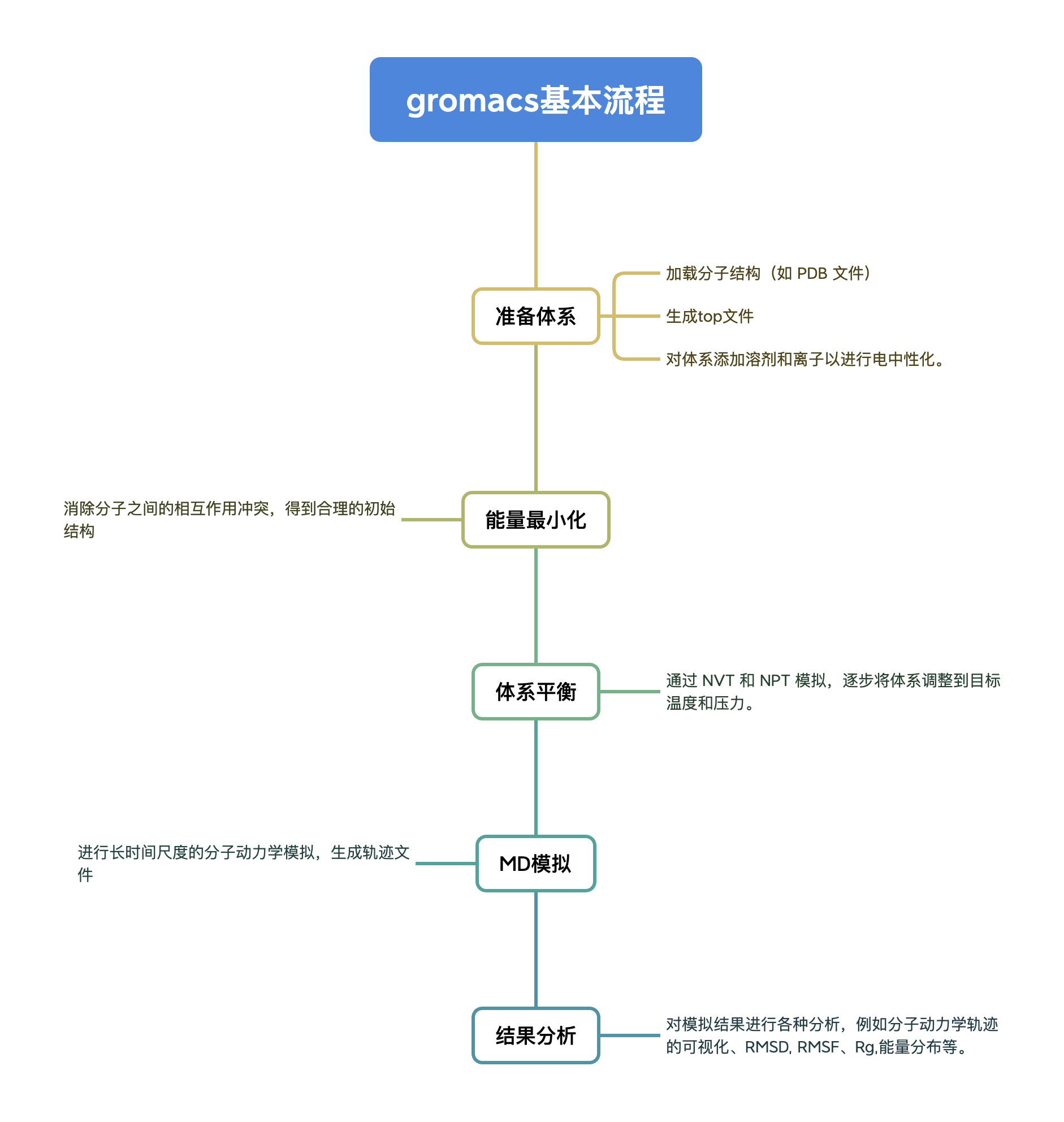
خطوات التشغيل
أولاً، نحتاج إلى تكوين ملف بروتين pdb وملف حساب md، ثم بعد المعالجة المسبقة المختلفة، نقوم أخيرًا بإجراء محاكاة 10ns وتحليل النتائج. وفيما يلي شرح خطوة بخطوة.
1. ابدأ GROMACS
首先登录平台:https://openbayes.com/
选择「高性能计算」> 创建新容器> 选择算力 RTX 4090> 选择 gromacs GPU
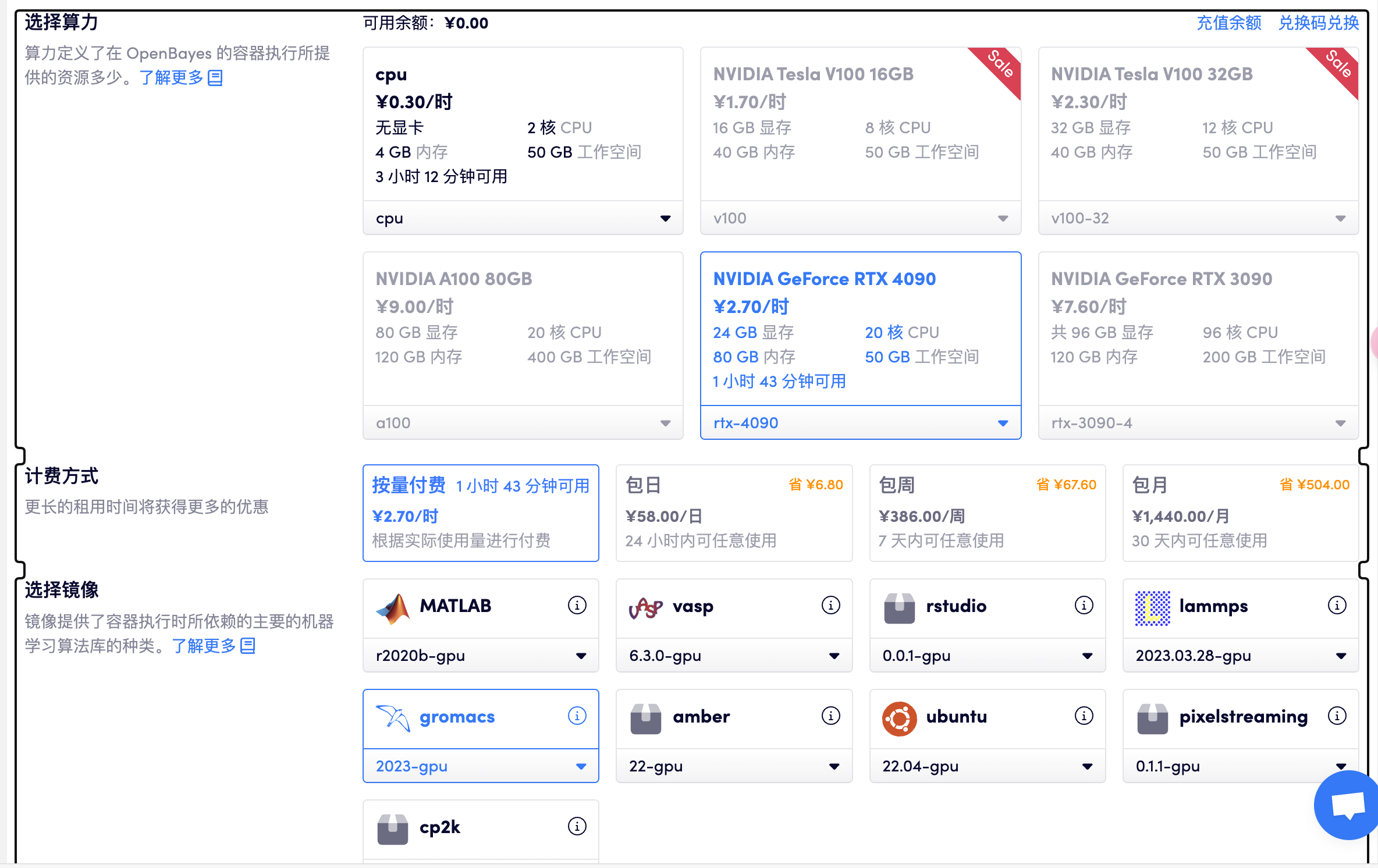
打开工作空间
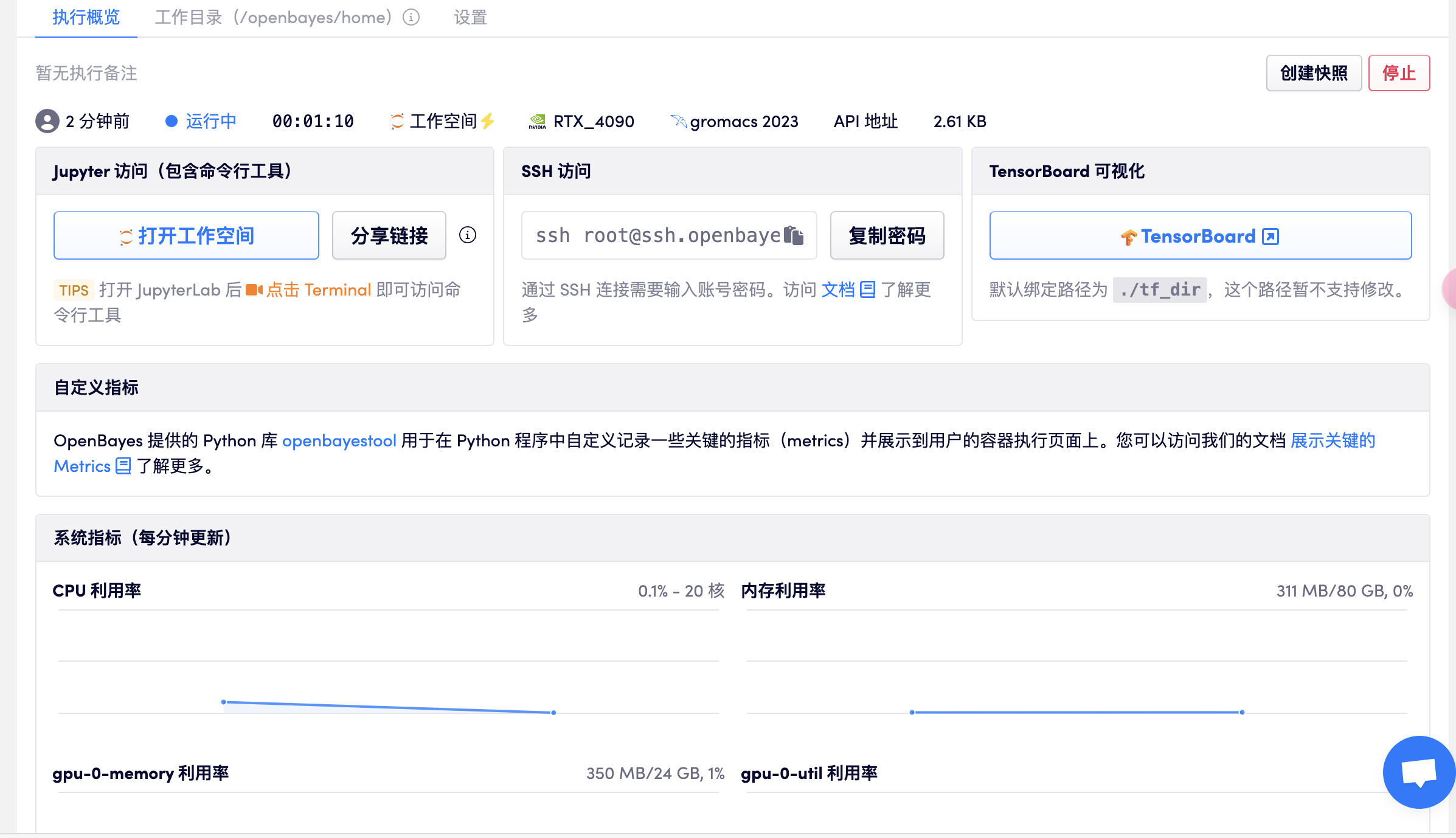
打开终端
或者采用 SSH 控制服务器:
在 X-shell 或者 mac unix 终端,输入:ssh -p 32699 [email protected],再输入密码即可(如下所示)↓
liangzhongzhongzhong@lzr ~ % ssh -p 32699 [email protected]
The authenticity of host '[ssh.openbayes.com]:32699 ([101.237.34.75]:32699)' can't be established.
ED25519 key fingerprint is SHA256:uwPyhP/EYoW49Ez4rvAuaf19czwis2rdS4pImsR0NH8.
This key is not known by any other names.
Are you sure you want to continue connecting (yes/no/[fingerprint])? yes
Warning: Permanently added '[ssh.openbayes.com]:32699' (ED25519) to the list of known hosts.
[email protected]'s password:
OpenBayes
目录说明
- /openbayes/home 工作空间内的数据保存地址,容器停止后,该目录中的内容不会被删除
- /openbayes/input/input0 - /openbayes/input/input4 为数据目录,不会占用工作空间的存储容量,最多支持同时绑定 5 个
⚠️ 其他目录下的内容在容器关闭后会被自动删除!更多信息请访问 https://openbayes.com/docs/concepts
⚠️ 禁止挖矿,一经发现将立即封号恕不退款
(base) root@liangzhong-4ay9ej85pxvd-main:/openbayes/home# ls
(base) root@liangzhong-4ay9ej85pxvd-main:/openbayes# l(base) root@liangzhong-4ay9ej85pxvd-main:/openbayes# lhome input 请将文件存在 home 目录下, 当前文件夹下的文件不会被保存.txt
(base) root@liangzhong-4ay9ej85pxvd-main:/openbayes# ls(base) root@liangzhong-4ay9ej85pxvd-main:/openbaye
(base) root@liangzhong-4ay9ej85pxvd-main:/openbayes/input# cd input0
(base) root@liangzhong-4ay9ej85pxvd-main:/openbayes/input/input0# ls
'#nvt.log.1#' '#topol.top.2#' 1AKI_processed.gro 1aki.pdb em.log ions.mdp mdout.mdp nvt.cpt nvt.log nvt.trr topol.top
'#nvt.log.2#' 1AKI_clean.pdb 1AKI_solv.gro em.edr em.tpr ions.tpr minim.mdp nvt.edr nvt.mdp posre.itp
'#topol.top.1#' 1AKI_newbox.gro 1AKI_solv_ions.gro em.gro em.trr md.mdp npt.mdp nvt.gro nvt.tpr potential.xvg
(base) root@liangzhong-4ay9ej85pxvd-main:/openbayes/input/input0#

调用 GROMACS,设置临时环境变量
export PATH=/data/app/gromacs/bin:$PATH
(base) root@liangzhong-4ay9ej85pxvd-main:/openbayes/home# gmx_mpi -h
:-) GROMACS - gmx_mpi, 2023 (-:
Executable: /data/app/gromacs/bin/gmx_mpi
Data prefix: /data/app/gromacs
Working dir: /output
Command line:
gmx_mpi -h
SYNOPSIS
gmx [-[no]h] [-[no]quiet] [-[no]version] [-[no]copyright] [-nice <int>]
[-[no]backup]
OPTIONS
Other options:
-[no]h (no)
Print help and quit
-[no]quiet (no)
Do not print common startup info or quotes
-[no]version (no)
Print extended version information and quit
-[no]copyright (no)
Print copyright information on startup
-nice <int> (19)
Set the nicelevel (default depends on command)
-[no]backup (yes)
Write backups if output files exist
Additional help is available on the following topics:
commands List of available commands
selections Selection syntax and usage
To access the help, use 'gmx help <topic>'.
For help on a command, use 'gmx help <command>'.
GROMACS reminds you: "All You Need is Greed" (Aztec Camera)
2. إعداد الوثائق
قبل تشغيل البرنامج التعليمي، نحتاج إلى تحضير الملفات الستة التالية: 1aki.pdb، ions.mdp، md.mdp، minim.mdp، npt.mdp، nvt.mdp
يمكن تنزيل هذه الملفات مباشرة، أو إنشاؤها على النحو التالي.
على سبيل المثال، ملف البروتين 1AKI.pdb: يمكن الحصول عليه من الكلية الملكية للجراحين في سانتا باربرا احصل على 1AKI.pdb من الموقع الإلكتروني.في البرنامج التعليمي، تم إنشاء ملفات أخرى عن طريق نسخ الكود باستخدام محرر النصوص vim.
مقدمة لقاعدة بيانات RCSB:
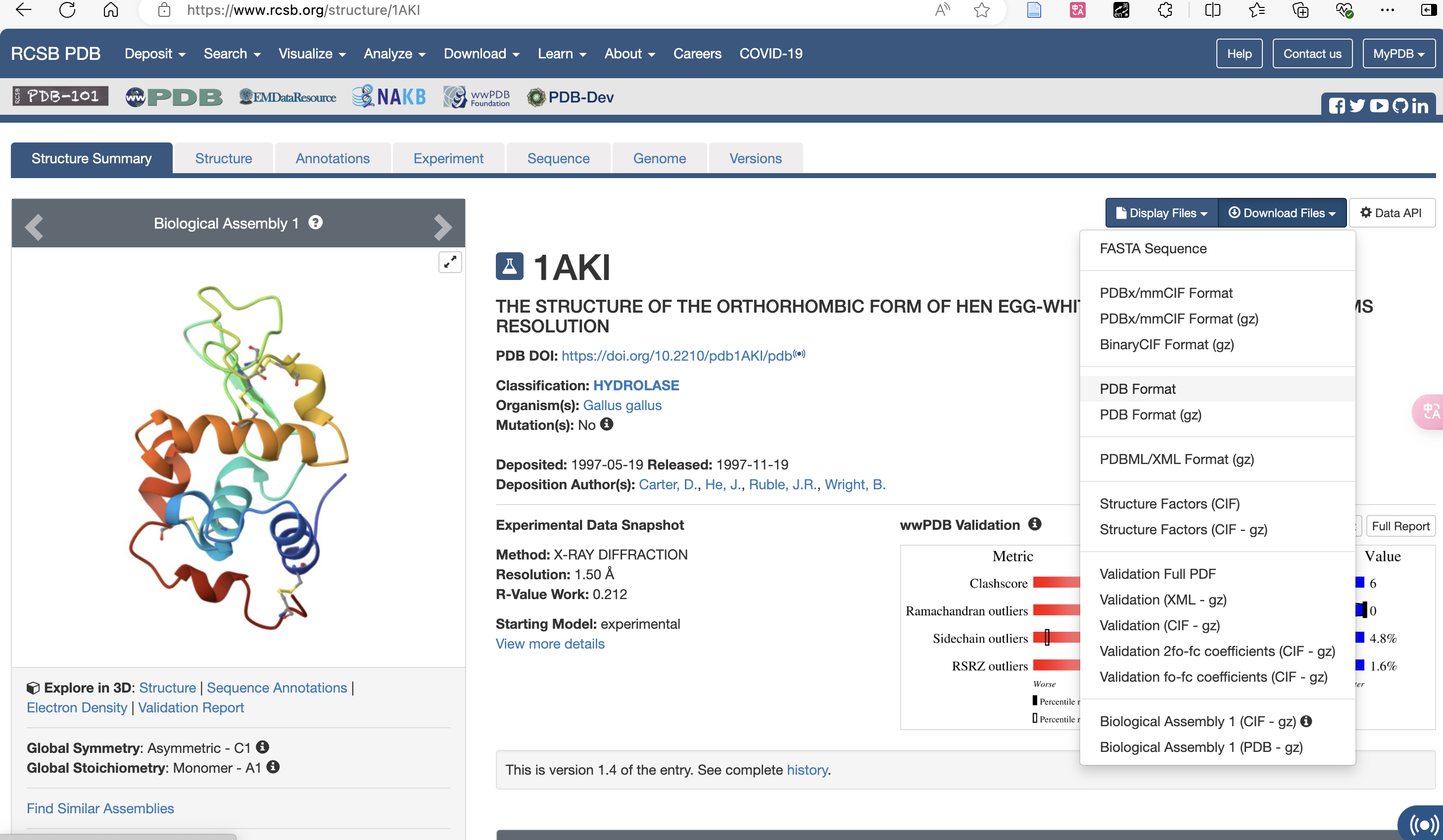
تنزيل تنسيق pdb:
تحميل إلى دليل العمل الخاص بك
(base) root@liangzhong-4ay9ej85pxvd-main:/input0# ls
1aki.pdb
#查看上传成功
vim ions.mdp
#准备 mdp 文件,输入命令后复制一下内容,按 i 进入插入模式开始编辑。
#编辑完成后,按 Esc 键退出插入模式,输入 :wq 保存并退出。
; ions.mdp - used as input into grompp to generate ions.tpr
; Parameters describing what to do, when to stop and what to save
integrator = steep ; Algorithm (steep = steepest descent minimization)
emtol = 1000.0 ; Stop minimization when the maximum force < 1000.0 kJ/mol/nm
emstep = 0.01 ; Minimization step size
nsteps = 50000 ; Maximum number of (minimization) steps to perform
; Parameters describing how to find the neighbors of each atom and how to calculate the interactions
nstlist = 1 ; Frequency to update the neighbor list and long range forces
cutoff-scheme = Verlet ; Buffered neighbor searching
ns_type = grid ; Method to determine neighbor list (simple, grid)
coulombtype = cutoff ; Treatment of long range electrostatic interactions
rcoulomb = 1.0 ; Short-range electrostatic cut-off
rvdw = 1.0 ; Short-range Van der Waals cut-off
pbc = xyz ; Periodic Boundary Conditions in all 3 dimensions
vim minim.mdp
; minim.mdp - used as input into grompp to generate em.tpr
; Parameters describing what to do, when to stop and what to save
integrator = steep ; Algorithm (steep = steepest descent minimization)
emtol = 1000.0 ; Stop minimization when the maximum force < 1000.0 kJ/mol/nm
emstep = 0.01 ; Minimization step size
nsteps = 50000 ; Maximum number of (minimization) steps to perform
; Parameters describing how to find the neighbors of each atom and how to calculate the interactions
nstlist = 1 ; Frequency to update the neighbor list and long range forces
cutoff-scheme = Verlet ; Buffered neighbor searching
ns_type = grid ; Method to determine neighbor list (simple, grid)
coulombtype = PME ; Treatment of long range electrostatic interactions
rcoulomb = 1.0 ; Short-range electrostatic cut-off
rvdw = 1.0 ; Short-range Van der Waals cut-off
pbc = xyz ; Periodic Boundary Conditions in all 3 dimensions
vim nvt.mdp
title = OPLS Lysozyme NVT equilibration
define = -DPOSRES ; position restrain the protein
; Run parameters
integrator = md ; leap-frog integrator
nsteps = 50000 ; 2 * 50000 = 100 ps
dt = 0.002 ; 2 fs
; Output control
nstxout = 500 ; save coordinates every 1.0 ps
nstvout = 500 ; save velocities every 1.0 ps
nstenergy = 500 ; save energies every 1.0 ps
nstlog = 500 ; update log file every 1.0 ps
; Bond parameters
continuation = no ; first dynamics run
constraint_algorithm = lincs ; holonomic constraints
constraints = h-bonds ; bonds involving H are constrained
lincs_iter = 1 ; accuracy of LINCS
lincs_order = 4 ; also related to accuracy
; Nonbonded settings
cutoff-scheme = Verlet ; Buffered neighbor searching
ns_type = grid ; search neighboring grid cells
nstlist = 10 ; 20 fs, largely irrelevant with Verlet
rcoulomb = 1.0 ; short-range electrostatic cutoff (in nm)
rvdw = 1.0 ; short-range van der Waals cutoff (in nm)
DispCorr = EnerPres ; account for cut-off vdW scheme
; Electrostatics
coulombtype = PME ; Particle Mesh Ewald for long-range electrostatics
pme_order = 4 ; cubic interpolation
fourierspacing = 0.16 ; grid spacing for FFT
; Temperature coupling is on
tcoupl = V-rescale ; modified Berendsen thermostat
tc-grps = Protein Non-Protein ; two coupling groups - more accurate
tau_t = 0.1 0.1 ; time constant, in ps
ref_t = 300 300 ; reference temperature, one for each group, in K
; Pressure coupling is off
pcoupl = no ; no pressure coupling in NVT
; Periodic boundary conditions
pbc = xyz ; 3-D PBC
; Velocity generation
gen_vel = yes ; assign velocities from Maxwell distribution
gen_temp = 300 ; temperature for Maxwell distribution
gen_seed = -1 ; generate a random seed
vim npt.mdp
title = OPLS Lysozyme NPT equilibration
define = -DPOSRES ; position restrain the protein
; Run parameters
integrator = md ; leap-frog integrator
nsteps = 50000 ; 2 * 50000 = 100 ps
dt = 0.002 ; 2 fs
; Output control
nstxout = 500 ; save coordinates every 1.0 ps
nstvout = 500 ; save velocities every 1.0 ps
nstenergy = 500 ; save energies every 1.0 ps
nstlog = 500 ; update log file every 1.0 ps
; Bond parameters
continuation = yes ; Restarting after NVT
constraint_algorithm = lincs ; holonomic constraints
constraints = h-bonds ; bonds involving H are constrained
lincs_iter = 1 ; accuracy of LINCS
lincs_order = 4 ; also related to accuracy
; Nonbonded settings
cutoff-scheme = Verlet ; Buffered neighbor searching
ns_type = grid ; search neighboring grid cells
nstlist = 10 ; 20 fs, largely irrelevant with Verlet scheme
rcoulomb = 1.0 ; short-range electrostatic cutoff (in nm)
rvdw = 1.0 ; short-range van der Waals cutoff (in nm)
DispCorr = EnerPres ; account for cut-off vdW scheme
; Electrostatics
coulombtype = PME ; Particle Mesh Ewald for long-range electrostatics
pme_order = 4 ; cubic interpolation
fourierspacing = 0.16 ; grid spacing for FFT
; Temperature coupling is on
tcoupl = V-rescale ; modified Berendsen thermostat
tc-grps = Protein Non-Protein ; two coupling groups - more accurate
tau_t = 0.1 0.1 ; time constant, in ps
ref_t = 300 300 ; reference temperature, one for each group, in K
; Pressure coupling is on
pcoupl = Parrinello-Rahman ; Pressure coupling on in NPT
pcoupltype = isotropic ; uniform scaling of box vectors
tau_p = 2.0 ; time constant, in ps
ref_p = 1.0 ; reference pressure, in bar
compressibility = 4.5e-5 ; isothermal compressibility of water, bar^-1
refcoord_scaling = com
; Periodic boundary conditions
pbc = xyz ; 3-D PBC
; Velocity generation
gen_vel = no ; Velocity generation is off
vim md.md
title = OPLS Lysozyme NPT equilibration
; Run parameters
integrator = md ; leap-frog integrator
nsteps = 500000 ; 2 * 500000 = 1000 ps (1 ns)
dt = 0.002 ; 2 fs
; Output control
nstxout = 0 ; suppress bulky .trr file by specifying
nstvout = 0 ; 0 for output frequency of nstxout,
nstfout = 0 ; nstvout, and nstfout
nstenergy = 5000 ; save energies every 10.0 ps
nstlog = 5000 ; update log file every 10.0 ps
nstxout-compressed = 5000 ; save compressed coordinates every 10.0 ps
compressed-x-grps = System ; save the whole system
; Bond parameters
continuation = yes ; Restarting after NPT
constraint_algorithm = lincs ; holonomic constraints
constraints = h-bonds ; bonds involving H are constrained
lincs_iter = 1 ; accuracy of LINCS
lincs_order = 4 ; also related to accuracy
; Neighborsearching
cutoff-scheme = Verlet ; Buffered neighbor searching
ns_type = grid ; search neighboring grid cells
nstlist = 10 ; 20 fs, largely irrelevant with Verlet scheme
rcoulomb = 1.0 ; short-range electrostatic cutoff (in nm)
rvdw = 1.0 ; short-range van der Waals cutoff (in nm)
; Electrostatics
coulombtype = PME ; Particle Mesh Ewald for long-range electrostatics
pme_order = 4 ; cubic interpolation
fourierspacing = 0.16 ; grid spacing for FFT
; Temperature coupling is on
tcoupl = V-rescale ; modified Berendsen thermostat
tc-grps = Protein Non-Protein ; two coupling groups - more accurate
tau_t = 0.1 0.1 ; time constant, in ps
ref_t = 300 300 ; reference temperature, one for each group, in K
; Pressure coupling is on
pcoupl = Parrinello-Rahman ; Pressure coupling on in NPT
pcoupltype = isotropic ; uniform scaling of box vectors
tau_p = 2.0 ; time constant, in ps
ref_p = 1.0 ; reference pressure, in bar
compressibility = 4.5e-5 ; isothermal compressibility of water, bar^-1
; Periodic boundary conditions
pbc = xyz ; 3-D PBC
; Dispersion correction
DispCorr = EnerPres ; account for cut-off vdW scheme
; Velocity generation
gen_vel = no ; Velocity generation is off
2. خطوات المحاكاة الرسمية
هدف:بناء نظام محاكاة معقول من الناحية الفيزيائية لوضع الأساس للمحاكاة الديناميكية اللاحقة.
1. تحميل البنية الجزيئية (ملف PDB):
• ملفات PDB(بنك بيانات البروتين) يحتوي على إحداثيات ثلاثية الأبعاد ومعلومات بنيوية للجزيئات (مثل البروتينات والأحماض النووية والجزيئات الصغيرة).
• يستخدم GROMACS أداة pdb2gmx لتحويل معلومات الإحداثيات هذه إلى نموذج جزيئي وتعيين معلمات مجال القوة.
• مجال القوة:نموذج رياضي يصف التفاعلات بين الجزيئات، بما في ذلك الروابط، والزوايا، والطاقة الكامنة ثنائية السطوح، وقوى فان دير فالس، وتفاعلات الشحنة.
• حقول القوة المشتركة: GROMOS، AMBER، CHARMM.
(الأكثر استخدامًا هي بروتين AMBER99SB، وحقل القوة الذري OPLS-AA/L، وحقل القوة charm36 (يجب تنزيله وتكوينه بنفسك)، وحقول القوة الأخرى قديمة جدًا وغير مناسبة لنشر المقالات)
2. إنشاء ملف الطوبولوجيا:
• يحدد ملف الطوبولوجيا (topol.top) معلمات مجال القوة لكل جزيء في نظام المحاكاة، مثل نوع الذرة ونوع الرابطة ومعامالاتها، وما إلى ذلك.
• يتم دمجه مع ملف الإحداثيات لتشكيل أساس لمحاكاة الديناميكيات الجزيئية.
3. قم بتشغيل pdb2gmx، وحدد حقل القوة: 15 واضغط على Enter
#删除水分子(PDB 文件中的 “HOH” 残基)
grep -v HOH 1aki.pdb > 1AKI_clean.pdb
gmx_mpi pdb2gmx -f 1AKI_clean.pdb -o 1AKI_processed.gro -water spce
#选择 15,按回车,OPLS-AA/L all-atom force field (2001 aminoacid dihedrals)
(base) root@liangzhong-4ay9ej85pxvd-main:/input0# pdb2gmx -f 1AKI_clean.pdb -o 1AKI_processed.gro -water spce
:-) GROMACS - gmx pdb2gmx, 2023 (-:
Executable: /data/app/gromacs/bin/gmx_mpi
Data prefix: /data/app/gromacs
Working dir: /input0
Command line:
gmx_mpi pdb2gmx -f 1AKI_clean.pdb -o 1AKI_processed.gro -water spce
Select the Force Field:
From '/data/app/gromacs/share/gromacs/top':
1: AMBER03 protein, nucleic AMBER94 (Duan et al., J. Comp. Chem. 24, 1999-2012, 2003)
2: AMBER94 force field (Cornell et al., JACS 117, 5179-5197, 1995)
3: AMBER96 protein, nucleic AMBER94 (Kollman et al., Acc. Chem. Res. 29, 461-469, 1996)
4: AMBER99 protein, nucleic AMBER94 (Wang et al., J. Comp. Chem. 21, 1049-1074, 2000)
5: AMBER99SB protein, nucleic AMBER94 (Hornak et al., Proteins 65, 712-725, 2006)
6: AMBER99SB-ILDN protein, nucleic AMBER94 (Lindorff-Larsen et al., Proteins 78, 1950-58, 2010)
7: AMBERGS force field (Garcia & Sanbonmatsu, PNAS 99, 2782-2787, 2002)
8: CHARMM27 all-atom force field (CHARM22 plus CMAP for proteins)
9: GROMOS96 43a1 force field
10: GROMOS96 43a2 force field (improved alkane dihedrals)
11: GROMOS96 45a3 force field (Schuler JCC 2001 22 1205)
12: GROMOS96 53a5 force field (JCC 2004 vol 25 pag 1656)
13: GROMOS96 53a6 force field (JCC 2004 vol 25 pag 1656)
14: GROMOS96 54a7 force field (Eur. Biophys. J. (2011), 40,, 843-856, DOI: 10.1007/s00249-011-0700-9)
15: OPLS-AA/L all-atom force field (2001 aminoacid dihedrals)
15
Using the Oplsaa force field in directory oplsaa.ff
going to rename oplsaa.ff/aminoacids.r2b
Opening force field file /data/app/gromacs/share/gromacs/top/oplsaa.ff/aminoacids.r2b
Reading 1AKI_clean.pdb...
WARNING: all CONECT records are ignored
Read 'LYSOZYME', 1001 atoms
Analyzing pdb file
Splitting chemical chains based on TER records or chain id changing.
There are 1 chains and 0 blocks of water and 129 residues with 1001 atoms
chain #res #atoms
1 'A' 129 1001
All occupancies are one
All occupancies are one
Opening force field file /data/app/gromacs/share/gromacs/top/oplsaa.ff/atomtypes.atp
Reading residue database... (Oplsaa)
Opening force field file /data/app/gromacs/share/gromacs/top/oplsaa.ff/aminoacids.rtp
Opening force field file /data/app/gromacs/share/gromacs/top/oplsaa.ff/aminoacids.hdb
Opening force field file /data/app/gromacs/share/gromacs/top/oplsaa.ff/aminoacids.n.tdb
Opening force field file /data/app/gromacs/share/gromacs/top/oplsaa.ff/aminoacids.c.tdb
Processing chain 1 'A' (1001 atoms, 129 residues)
Analysing hydrogen-bonding network for automated assignment of histidine
protonation. 213 donors and 184 acceptors were found.
There are 255 hydrogen bonds
Will use HISE for residue 15
Identified residue LYS1 as a starting terminus.
Identified residue LEU129 as a ending terminus.
8 out of 8 lines of specbond.dat converted successfully
Special Atom Distance matrix:
CYS6 MET12 HIS15 CYS30 CYS64 CYS76 CYS80
SG48 SD87 NE2118 SG238 SG513 SG601 SG630
MET12 SD87 1.166
HIS15 NE2118 1.776 1.019
CYS30 SG238 1.406 1.054 2.069
CYS64 SG513 2.835 1.794 1.789 2.241
CYS76 SG601 2.704 1.551 1.468 2.116 0.765
CYS80 SG630 2.959 1.951 1.916 2.391 0.199 0.944
CYS94 SG724 2.550 1.407 1.382 1.975 0.665 0.202 0.855
MET105 SD799 1.827 0.911 1.683 0.888 1.849 1.461 2.036
CYS115 SG889 1.576 1.084 2.078 0.200 2.111 1.989 2.262
CYS127 SG981 0.197 1.072 1.721 1.313 2.799 2.622 2.934
CYS94 MET105 CYS115
SG724 SD799 SG889
MET105 SD799 1.381
CYS115 SG889 1.853 0.790
CYS127 SG981 2.475 1.686 1.483
Linking CYS-6 SG-48 and CYS-127 SG-981...
Linking CYS-30 SG-238 and CYS-115 SG-889...
Linking CYS-64 SG-513 and CYS-80 SG-630...
Linking CYS-76 SG-601 and CYS-94 SG-724...
Start terminus LYS-1: NH3+
End terminus LEU-129: COO-
Checking for duplicate atoms....
Generating any missing hydrogen atoms and/or adding termini.
Now there are 129 residues with 1960 atoms
Making bonds...
Number of bonds was 1984, now 1984
Generating angles, dihedrals and pairs...
Before cleaning: 5142 pairs
Before cleaning: 5187 dihedrals
Making cmap torsions...
There are 5187 dihedrals, 426 impropers, 3547 angles
5106 pairs, 1984 bonds and 0 virtual sites
Total mass 14313.193 a.m.u.
Total charge 8.000 e
Writing topology
Writing coordinate file...
--------- PLEASE NOTE ------------
You have successfully generated a topology from: 1AKI_clean.pdb.
The Oplsaa force field and the spce water model are used.
--------- ETON ESAELP ------------
GROMACS reminds you: "Any one who considers arithmetical methods of producing random digits is, of course, in a state of sin." (John von Neumann)
يمكنك أن ترى أن هناك العديد من الملفات الأخرى، و"إجمالي الرسوم 8.000 جنيه إسترليني" سوف نحتاج إلى تحييد الرسوم لاحقًا
استخدم الأمر vim لعرض topol.top ويمكنك رؤية علامة حقل القوة.
(base) root@liangzhong-4ay9ej85pxvd-main:/input0# ls
1AKI_clean.pdb 1aki.pdb md.mdp npt.mdp posre.itp
1AKI_processed.gro ions.mdp minim.mdp nvt.mdp topol.top
4. قم بإنشاء صندوق معيني ذي اثني عشر وجهًا لإغلاق البروتين حتى نتمكن من إضافة جزيئات المذيب
• اختيار شكل الصندوق:تتضمن أشكال صناديق المحاكاة الشائعة المكعب، والصندوق المتعامد، والمعين الاثني عشري الوجوه، وما إلى ذلك. وعادةً ما يتم اختيار الأشكال التي يمكنها ملء الجزيئات بشكل فعال وتقليل كمية الحساب.
gmx_mpi editconf -f 1AKI_processed.gro -o 1AKI_newbox.gro -c -d 1.0 -bt cubic
#将蛋白质在框中居中(-c),并将其放置在框边缘至少 1.0 nm 的位置(-d 1.0)
Command line:
gmx_mpi editconf -f 1AKI_processed.gro -o 1AKI_newbox.gro -c -d 1.0 -bt cubic
Note that major changes are planned in future for editconf, to improve usability and utility.
Read 1960 atoms
Volume: 123.376 nm^3, corresponds to roughly 55500 electrons
No velocities found
system size : 3.817 4.234 3.454 (nm)
diameter : 5.010 (nm)
center : 2.781 2.488 0.017 (nm)
box vectors : 5.906 6.845 3.052 (nm)
box angles : 90.00 90.00 90.00 (degrees)
box volume : 123.38 (nm^3)
shift : 0.724 1.017 3.488 (nm)
new center : 3.505 3.505 3.505 (nm)
new box vectors : 7.010 7.010 7.010 (nm)
new box angles : 90.00 90.00 90.00 (degrees)
new box volume : 344.48 (nm^3)
5. حدد مربعًا وأضف المذيب (الماء)
غاية:المذيب:ضع الجزيئات في بيئة مذيبة (مثل صندوق ماء) لمحاكاة سلوكها في البيئة الحقيقية.
gmx_mpi solvate -cp 1AKI_newbox.gro -cs spc216.gro -o 1AKI_solv.gro -p topol.top
#结果
Generating solvent configuration
Will generate new solvent configuration of 4x4x4 boxes
Solvent box contains 39252 atoms in 13084 residues
Removed 5451 solvent atoms due to solvent-solvent overlap
Removed 1869 solvent atoms due to solute-solvent overlap
Sorting configuration
Found 1 molecule type:
SOL ( 3 atoms): 10644 residues
Generated solvent containing 31932 atoms in 10644 residues
Writing generated configuration to 1AKI_solv.gro
Output configuration contains 33892 atoms in 10773 residues
Volume : 344.484 (nm^3)
Density : 997.935 (g/l)
Number of solvent molecules: 10644
Processing topology
Adding line for 10644 solvent molecules with resname (SOL) to topology file (topol.top)
#查看一下 top 文件
(base) root@liangzhong-4ay9ej85pxvd-main:/input0# ls
'#topol.top.1#' 1AKI_newbox.gro 1AKI_solv.gro ions.mdp minim.mdp nvt.mdp topol.top
1AKI_clean.pdb 1AKI_processed.gro 1aki.pdb md.mdp npt.mdp posre.itp
(base) root@liangzhong-4ay9ej85pxvd-main:/input0# vim topol.top
#查看 top 文件的末尾
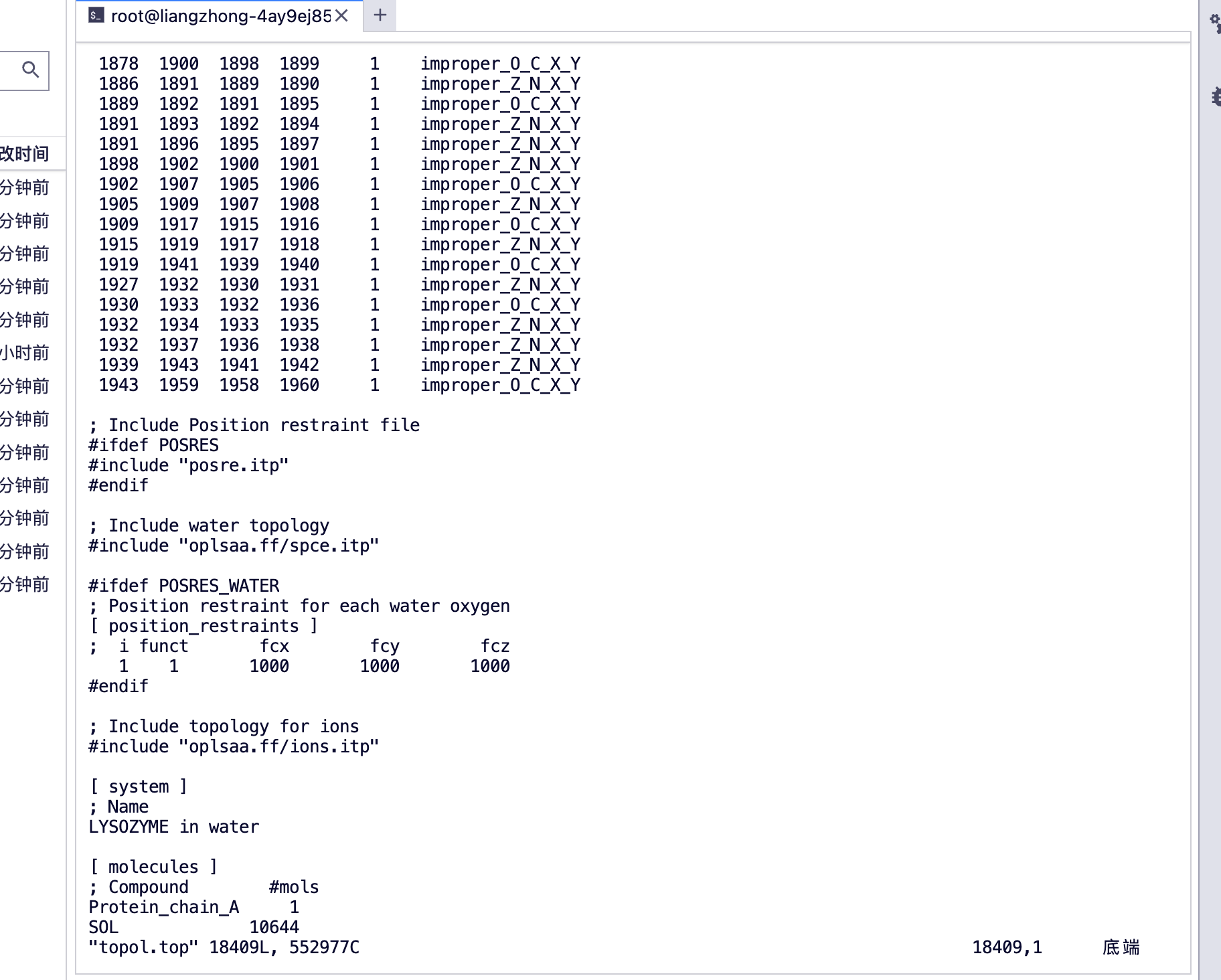
يمكنك رؤية [الجزيئات]
سلسلة البروتين أ، سلسلة البروتين أ
جزيء الماء SOL
6. قم بتجميع ملف .tpr
gmx_mpi grompp -f ions.mdp -c 1AKI_solv.gro -p topol.top -o ions.tpr
7. تحييد الشحنة واستبدال جزيئات المذيب بالأيونات
تحييد الشحنة:أضف أيونات (مثل Na⁺ وCl⁻) لتحييد الشحنة الكلية للنظام وتجنب تشوه المحاكاة الناجم عن التأثيرات الكهروستاتيكية.
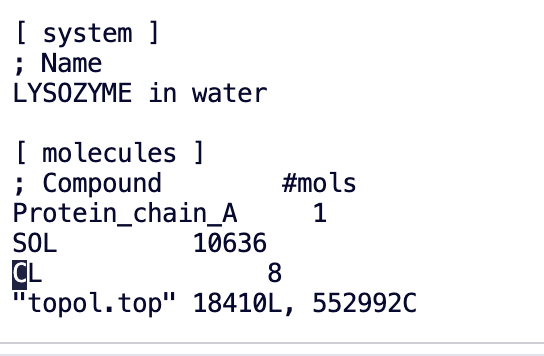
gmx_mpi genion -s ions.tpr -o 1AKI_solv_ions.gro -p topol.top -pname NA -nname CL -neutral
Command line:
gmx_mpi genion -s ions.tpr -o 1AKI_solv_ions.gro -p topol.top -pname NA -nname CL -neutral
Reading file ions.tpr, VERSION 2023 (single precision)
Reading file ions.tpr, VERSION 2023 (single precision)
Will try to add 0 NA ions and 8 CL ions.
Select a continuous group of solvent molecules
Group 0 ( System) has 33892 elements
Group 1 ( Protein) has 1960 elements
Group 2 ( Protein-H) has 1001 elements
Group 3 ( C-alpha) has 129 elements
Group 4 ( Backbone) has 387 elements
Group 5 ( MainChain) has 517 elements
Group 6 ( MainChain+Cb) has 634 elements
Group 7 ( MainChain+H) has 646 elements
Group 8 ( SideChain) has 1314 elements
Group 9 ( SideChain-H) has 484 elements
Group 10 ( Prot-Masses) has 1960 elements
Group 11 ( non-Protein) has 31932 elements
Group 12 ( Water) has 31932 elements
Group 13 ( SOL) has 31932 elements
Group 14 ( non-Water) has 1960 elements
Select a group: 13
Selected 13: 'SOL'
Number of (3-atomic) solvent molecules: 10644
Processing topology
Replacing 8 solute molecules in topology file (topol.top) by 0 NA and 8 CL ions.
Back Off! I just backed up topol.top to ./#topol.top.2#
Using random seed -1212354563.
Replacing solvent molecule 3671 (atom 12973) with CL
Replacing solvent molecule 2264 (atom 8752) with CL
Replacing solvent molecule 2559 (atom 9637) with CL
Replacing solvent molecule 8081 (atom 26203) with CL
Replacing solvent molecule 8468 (atom 27364) with CL
Replacing solvent molecule 7439 (atom 24277) with CL
Replacing solvent molecule 9983 (atom 31909) with CL
Replacing solvent molecule 650 (atom 3910) with CL
GROMACS reminds you: "Water is just water" (Berk Hess)
(base) root@liangzhong-4ay9ej85pxvd-main:/input0#
يمكن ملاحظة أنه تمت إضافة الكثير من أيونات الكلوريد
8. تقليل استهلاك الطاقة
8.1 السبب: عندما نجري محاكاة ديناميكية جزيئية، فإن ملفات قاعدة بيانات البروتين التي نحصل عليها يتم الحصول عليها من خلال الأشعة السينية والمجهر الإلكتروني وغيرها من الطرق، والتي تحتوي على العديد من زوايا الرابطة والتشوهات ذات الطاقة الزائدة، أو تكوين أو كسر الروابط الهيدروجينية، والترتيبات الخاصة بين الجزيئات، والطاقة العالية الناجمة عن المسافة القريبة بين الذرات، مما يجعل من الصعب على النظام الجزيئي في المحاكاة التحول من حالة عالية الطاقة إلى حالة منخفضة الطاقة. هذه الطريقة ضرورية لتقليل طاقة النظام.
8.2 المبدأ: المبدأ الأساسي لتقليل الطاقة: يعتمد تقليل الطاقة على دالة الطاقة الكامنة، والتي تقلل إجمالي الطاقة الكامنة للنظام عن طريق الحساب التكراري وتعديل إحداثيات الذرة. تتضمن الطرق المستخدمة بشكل شائع ما يلي:
① طريقة التدرج المترافق: طريقة الانحدار المتدرج التي تعمل على تسريع التقارب من خلال الاستفادة من المعلومات من الخطوة السابقة.
② طريقة الهبوط الأكثر انحدارًا: تتحرك كل تكرار في الاتجاه المعاكس لتدرج الطاقة الكامنة، مما يقلل الطاقة على طول مسار الهبوط الأكثر انحدارًا.
③طريقة نيوتن-رافسون: طريقة مشتقة من الدرجة الثانية تستخدم المشتقة من الدرجة الثانية لدالة الطاقة الكامنة للعثور على نقطة الطاقة الدنيا بدقة أكبر.
8.3 الغرض: إيجاد تكوين جزيئي أكثر استقرارًا عن طريق تعديل الإحداثيات الذرية للجزيء لتقليل الطاقة الكامنة الكلية للنظام.
① إزالة التكوينات الهندسية غير المعقولة: من خلال تقليل الطاقة، يمكن إزالة التكوينات الهندسية غير المعقولة في البنية الأولية، مثل التداخل الذري غير المعقول والتمدد، ويمكن القضاء على حالة الطاقة العالية التي تسببها، ويمكن جعل النظام أكثر استقرارًا.
② تحضير الهيكل الأولي: تأكد من أن الهيكل الأولي في حالة طاقة كامنة منخفضة لتجنب تأثير الطاقة العالية غير الضروري أثناء المحاكاة.
③ تحسين كفاءة الحوسبة: من خلال تقليل إجمالي الطاقة الكامنة للنظام، يمكن تحسين استقرار وكفاءة عمليات المحاكاة والحسابات اللاحقة
gmx_mpi grompp -f minim.mdp -c 1AKI_solv_ions.gro -p topol.top -o em.tpr
gmx_mpi mdrun -v -deffnm em

٩. ألقِ نظرة على النتائج (في نهاية البرنامج التعليمي، سنعلمك كيفية عرض ملفات xvg. يمكنك استخدام xmgrace وqtgrace وpython وexcel لإنشاء رسم بياني. وهو في الأساس رسم بياني خطي).
gmx_mpi energy -f em.edr -o potential.xvg
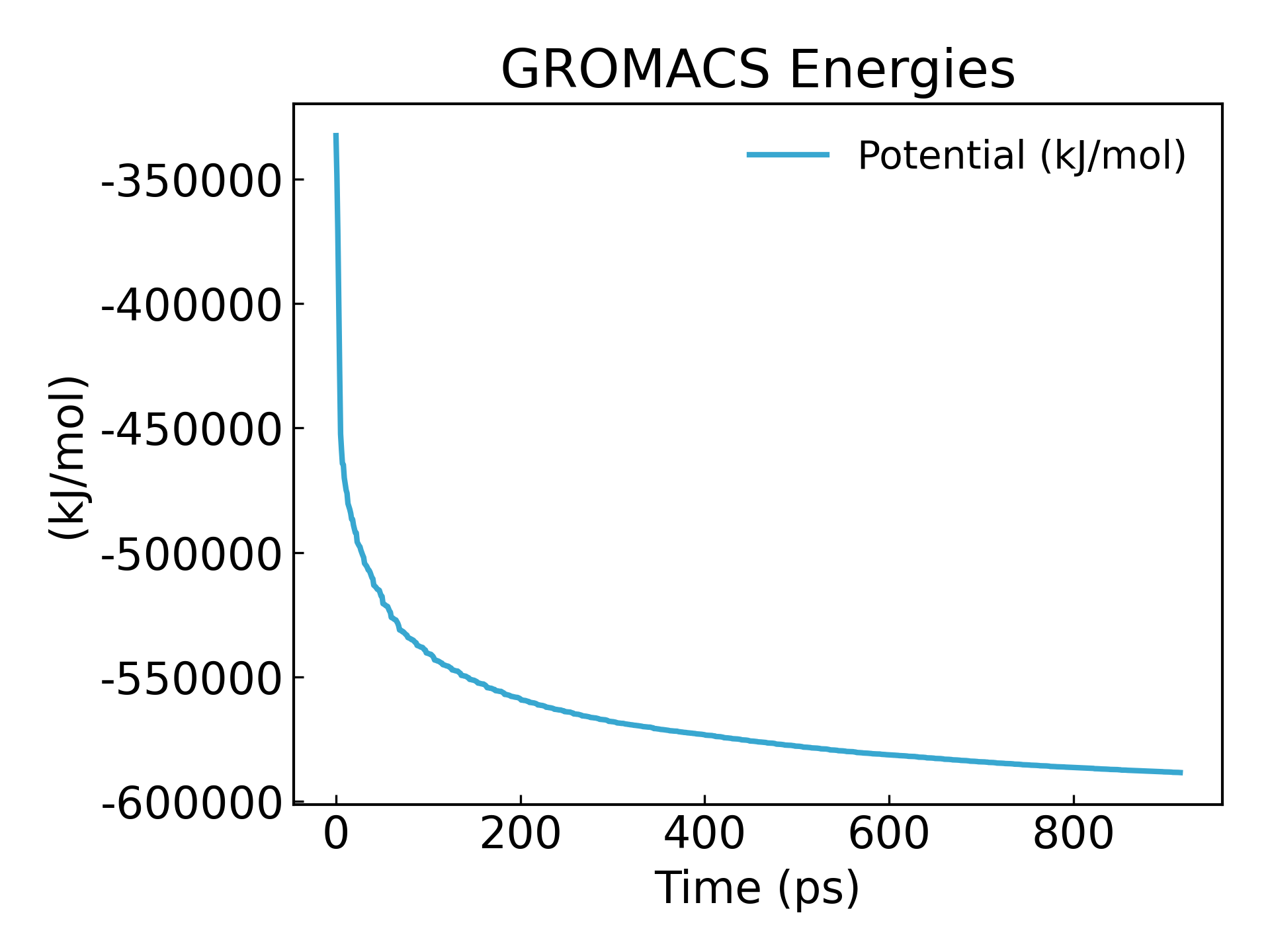
يمكن ملاحظة أن الطاقة تم تقليلها إلى -600000 كيلوجول/مول
10. توازن النظام
الهدف: ضبط النظام على درجة الحرارة والضغط المستهدفة لجعله قريبًا من الحالة الفيزيائية الحقيقية.
(1) محاكاة NVT (حجم ثابت ودرجة حرارة ثابتة):
• يتم استخدام مجموعة ذات حجم ثابت ودرجة حرارة ثابتة لتثبيت درجة حرارة النظام عند قيمة مستهدفة.
• وحدة التحكم في درجة الحرارة: يتم استخدام جهاز ربط درجة الحرارة Berendsen أو V-rescale (طريقة الربط الضعيف المعدلة) بشكل شائع، والذي يتحكم في درجة الحرارة عن طريق ضبط السرعة الذرية.
(2) محاكاة NPT (الضغط الثابت ودرجة الحرارة الثابتة):
• تعمل مجموعة الضغط الثابت ودرجة الحرارة الثابتة على ضبط كثافة النظام إلى قيمة مستهدفة (عادةً كثافة الماء السائل ~1 جم/سم³).
• وحدة التحكم في الضغط: يتم استخدام وصلة الضغط Berendsen أو طريقة التحكم في الضغط Parrinello-Rahman بشكل شائع.
10.1. إجراء موازنة NVT لـ 100 ps، والتي يتم إجراؤها تحت (عدد الجسيمات الثابت والحجم ودرجة الحرارة)، والمعروفة أيضًا باسم "المعادلة الحرارية والمتساوية الحرارة"
إنه سريع جدًا مع تسريع وحدة معالجة الرسوميات، إذ يستغرق 10 ثوانٍ أو أكثر فقط.
gmx_mpi grompp -f nvt.mdp -c em.gro -r em.gro -p topol.top -o nvt.tpr
gmx_mpi mdrun -deffnm nvt -nb gpu -pme cpu
#将 PME 任务移至 CPU

gmx_mpi energy -f nvt.edr -o temperature.xvg
#生成温度随时间变化的图像,查看温度是否平衡
16
0
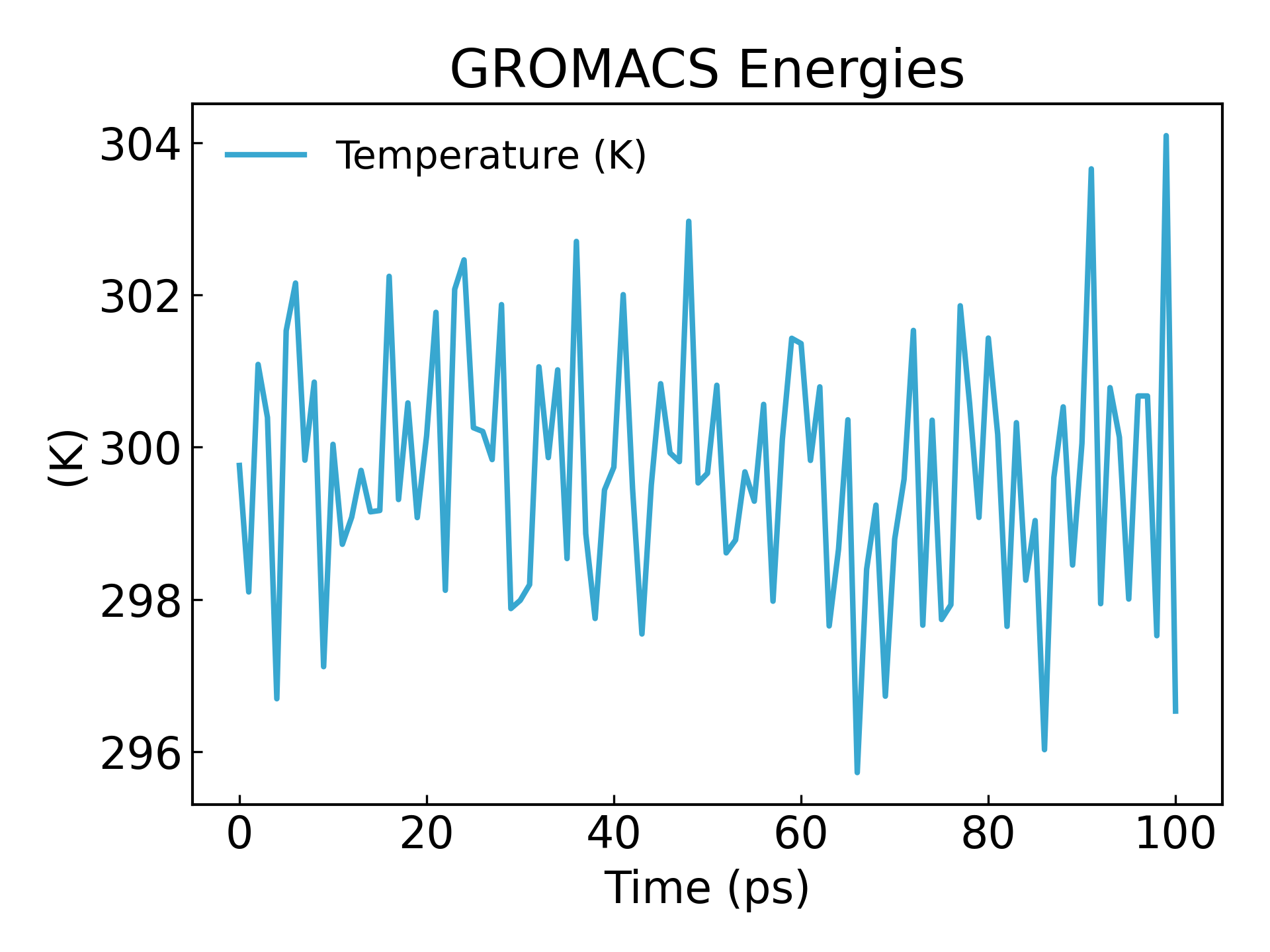
يمكن ملاحظة أن درجة الحرارة تصل أيضًا إلى حالة مستقرة أساسية في غضون 100 بيكو ثانية
10.2. تحقيق التوازن "المتساوي الحرارة والمتساوي الضغط" في نظام NPT، وتثبيت ضغط النظام، وإجراء توازن NPT 100 ps.
gmx_mpi grompp -f npt.mdp -c nvt.gro -r nvt.gro -t nvt.cpt -p topol.top -o npt.tpr
gmx_mpi mdrun -deffnm npt -nb gpu -pme cpu
#压力是否平衡
gmx_mpi energy -f npt.edr -o pressure.xvg
18
0
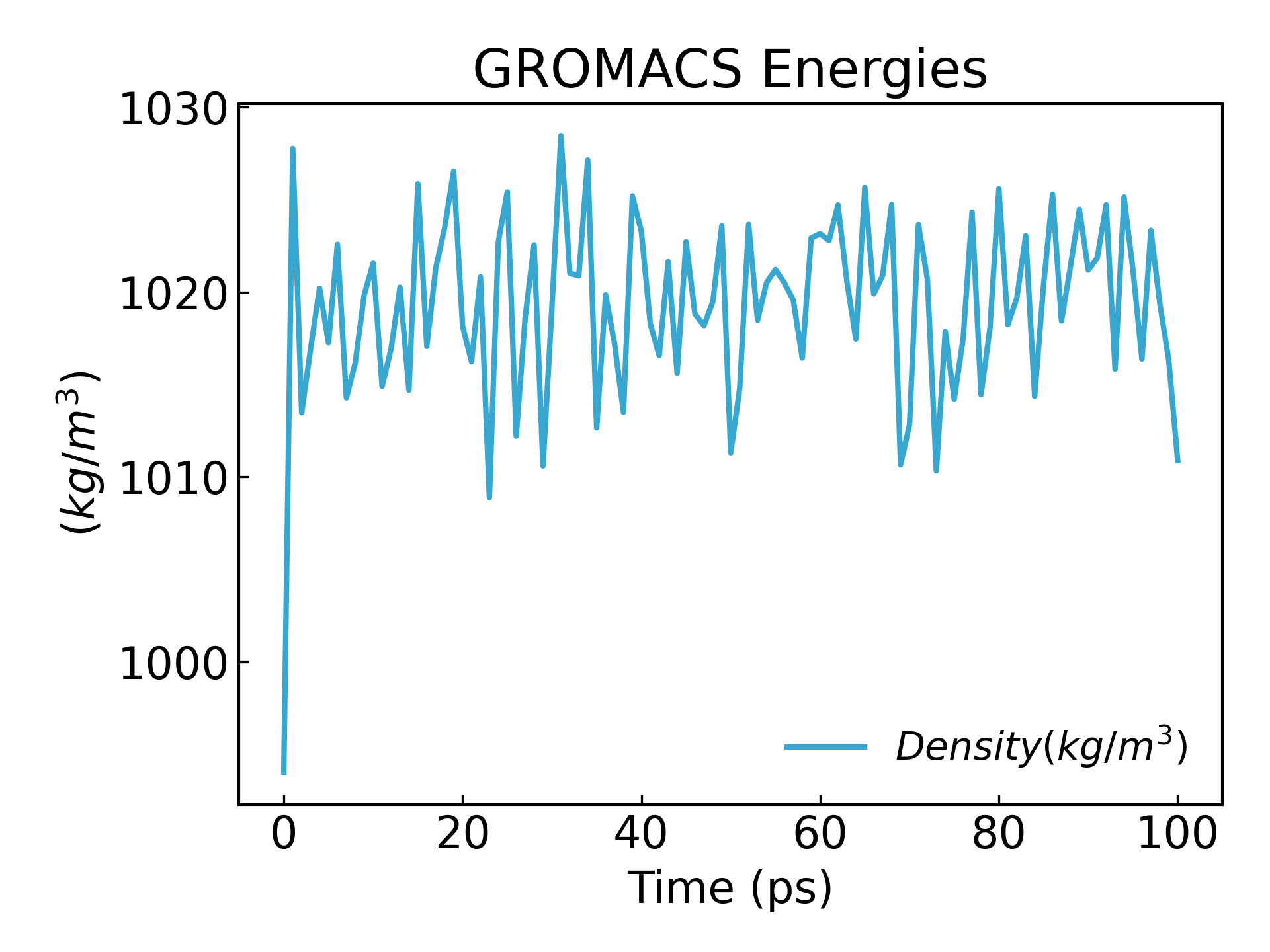
gmx_mpi energy -f npt.edr -o density.xvg
24
0
يمكننا أن نرى أن الكثافة تصل إلى حالة مستقرة:
11. بعد إكمال مرحلتي التوازن، أصبح النظام الآن متوازنًا جيدًا عند درجة الحرارة والضغط المطلوبين. يمكننا الآن تحرير قيود الموضع وتشغيل MD للمتابعة.
يمكنك تعديل الوقت حسب احتياجاتك. يحاكي هذا البرنامج التعليمي 100ns.
خطوة الوقت dt = 2 fs (الإعداد الشائع):
50000000 خطوة، بخطوة زمنية تبلغ 2 فمت ثانية، أي ما يعادل 100 نانوثانية (ns)
50000000 x2 fs = 10^8 fs = 10^5 ps =100ns
vim md.mdp
nsteps = 50000000 ; 2 * 50000000 = 100000 ps (100 ns)

gmx_mpi grompp -f md.mdp -c npt.gro -t npt.cpt -p topol.top -o md_0_1.tpr
##最后一步提交,pme 分配到 CPU 上:
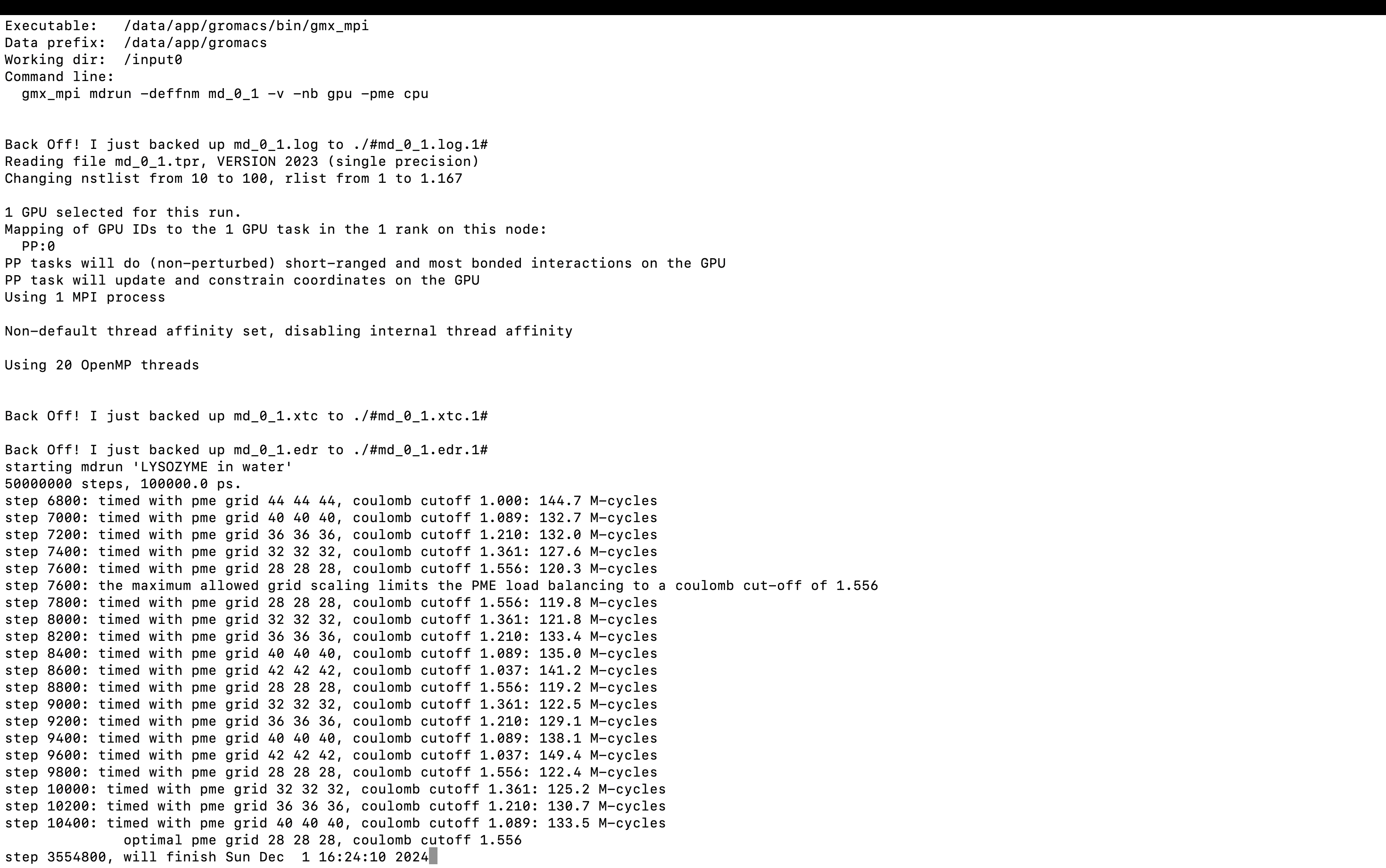
gmx_mpi mdrun -deffnm md_0_1 -v -nb gpu -pme cpu
-v يمكنه عرض الوقت المتبقي من التشغيل.
3. تحليل النتائج
1. trjconv، والذي يستخدم كأداة معالجة لاحقة لإزالة الإحداثيات أو تصحيح الدورية أو تعديل المسار يدويًا (وحدة الوقت، معدل الإطارات، وما إلى ذلك). ويرجع ذلك إلى أنه في جميع عمليات المحاكاة ذات الظروف الحدودية الدورية، قد تنكسر الجزيئات أو تقفز حول حافة الصندوق. يمكنه إعادة مركز الجزيء داخل الصندوق، وإعادة لف الجزيء، واستعادة الشكل المعيني الاثني عشري للصندوق.
-pbc mol: 去除轨迹中的周期性边界条件 (PBC),并基于分子进行修正(即确保每个分子在轨迹中是完整的)。
• -center: 将选定的分子/分组居中到模拟框的中心。
gmx_mpi trjconv -s md_0_1.tpr -f md_0_1.xtc -o md_0_1_noPBC.xtc -pbc mol -center
Select group for centering
Group 0 ( System) has 33876 elements
Group 1 ( Protein) has 1960 elements
Group 2 ( Protein-H) has 1001 elements
Group 3 ( C-alpha) has 129 elements
Group 4 ( Backbone) has 387 elements
Group 5 ( MainChain) has 517 elements
Group 6 ( MainChain+Cb) has 634 elements
Group 7 ( MainChain+H) has 646 elements
Group 8 ( SideChain) has 1314 elements
Group 9 ( SideChain-H) has 484 elements
Group 10 ( Prot-Masses) has 1960 elements
Group 11 ( non-Protein) has 31916 elements
Group 12 ( Water) has 31908 elements
Group 13 ( SOL) has 31908 elements
Group 14 ( non-Water) has 1968 elements
Group 15 ( Ion) has 8 elements
Group 16 ( Water_and_ions) has 31916 elements
Select a group: 1
Selected 1: 'Protein'
Select group for output
Group 0 ( System) has 33876 elements
Group 1 ( Protein) has 1960 elements
Group 2 ( Protein-H) has 1001 elements
Group 3 ( C-alpha) has 129 elements
Group 4 ( Backbone) has 387 elements
Group 5 ( MainChain) has 517 elements
Group 6 ( MainChain+Cb) has 634 elements
Group 7 ( MainChain+H) has 646 elements
Group 8 ( SideChain) has 1314 elements
Group 9 ( SideChain-H) has 484 elements
Group 10 ( Prot-Masses) has 1960 elements
Group 11 ( non-Protein) has 31916 elements
Group 12 ( Water) has 31908 elements
Group 13 ( SOL) has 31908 elements
Group 14 ( non-Water) has 1968 elements
Group 15 ( Ion) has 8 elements
Group 16 ( Water_and_ions) has 31916 elements
Select a group: 0
Selected 0: 'System'
تم استخدام الاختيار 4 ("العمود الفقري") لملاءمة المربعات الصغرى وحسابات RMSD.
2.RMSD
يمكن استخدامه للتحقق من تقارب المحاكاة واستقرار البروتين.g_rms**غالبًا ما يتم استخدام الانحراف المعياري الجذري المتوسط للهيكل أثناء المحاكاة والهيكل الأولي، وانحراف الهيكل في لحظة معينة بالنسبة إلى 0ns، لتقييم استقرار بنية البروتين. يتم استخدامه غالبًا في مجال تصميم الأدوية، مثل استقرار بنية ربيطة البروتين. إن نطاق التقلب لمنحنى RMSD صغير ومستقر، مما يشير إلى أن التقارب بين الربيطة والمستقبل كبير.
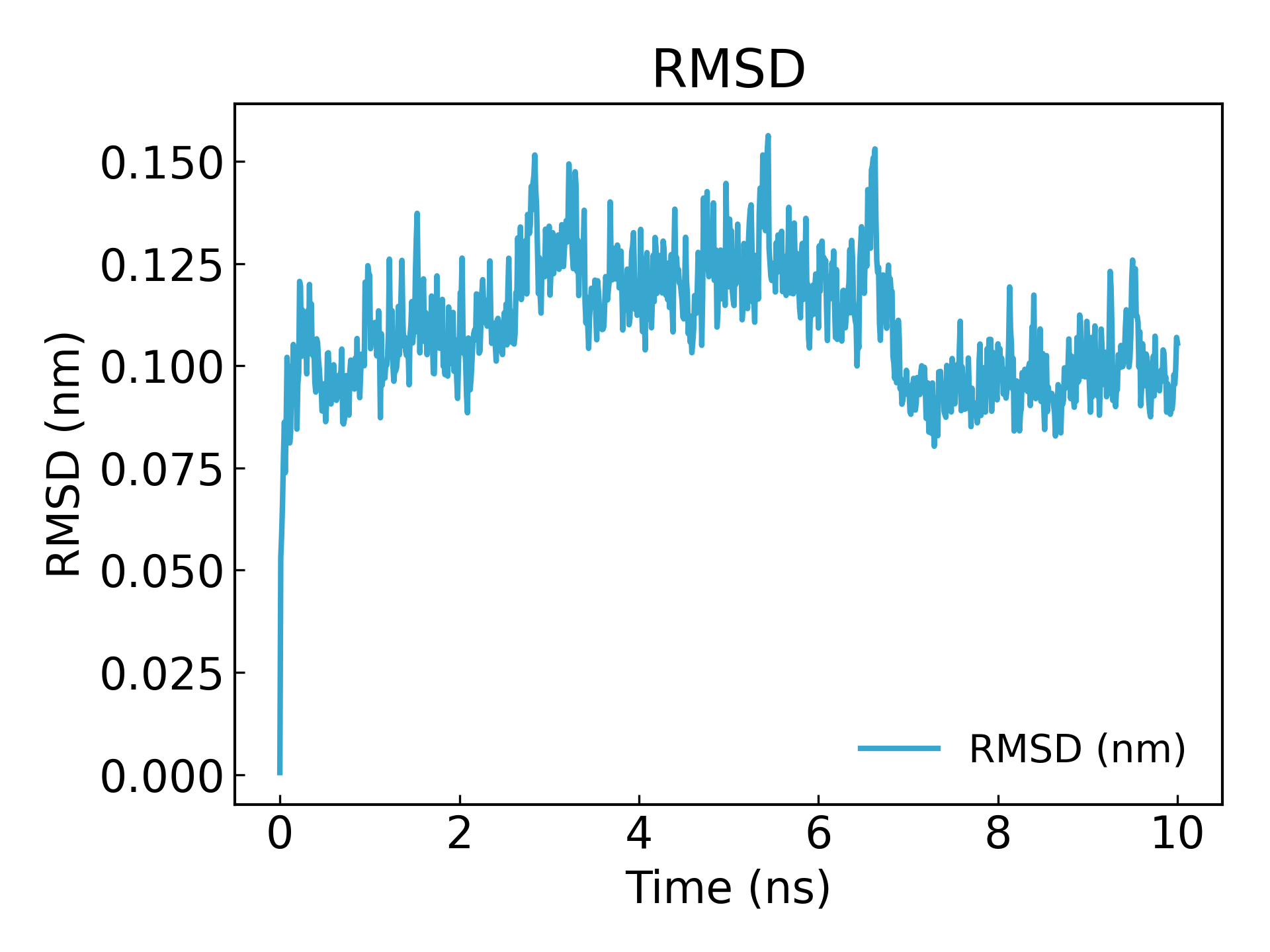
gmx_mpi rms -s md_0_1.tpr -f md_0_1_noPBC.xtc -o rmsd.xvg
حدد 4 ("العمود الفقري") لملاءمة المربعات الصغرى وحساب RMSD
يمكن ملاحظة أن التوازن يتم الوصول إليه بعد 10ns (ملاحظة: عند نشر أو إرسال مقال، من الأفضل المحاكاة لمدة 100ns أو 50ns)
3. تحليل نصف قطر الدوران
gmx_mpi gyrate -s md_0_1.tpr -f md_0_1_noPBC.xtc -o gyrate.xvg
نصف قطر دوران البروتين هو مقياس لمدى تماسكه. إذا كان هيكل البروتين مستقرًا، فمن المرجح أن يحافظ على قيمة Rg مستقرة نسبيًا. إذا انكشف البروتين، فإن قيمة Rg الخاصة به سوف تتغير مع مرور الوقت. دعونا نحلل نصف قطر دوران الليزوزيم في محاكاتنا:
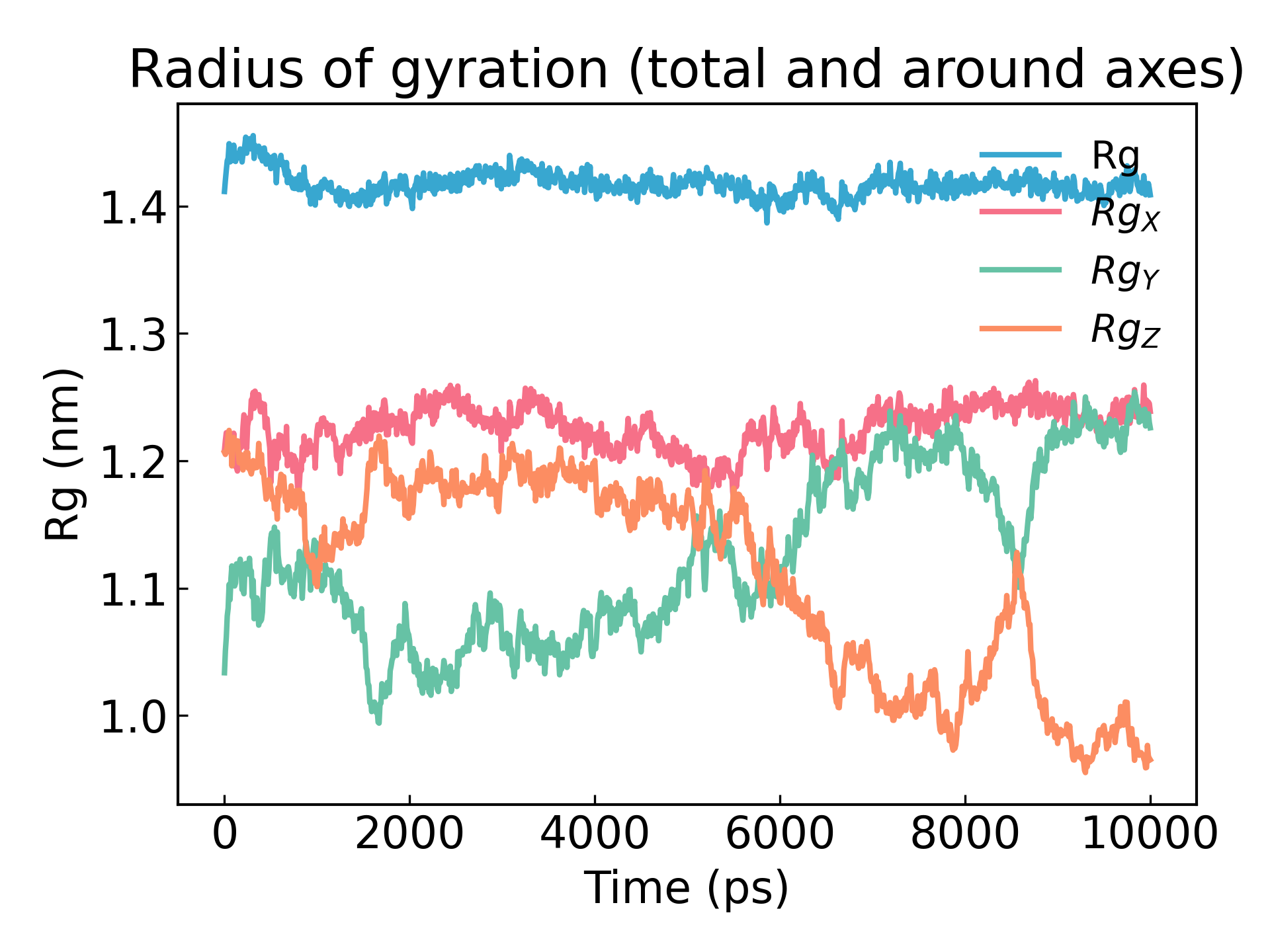
4. التصور
البرامج الموصى بها:DuIvyTools: أدوات تحليل وتصور محاكاة GROMACS
من:https://github.com/CharlesHahn/DuIvyTools
pip install DuIvyTools
dit xvg_show -f rmsd.xvg -o rmsd_plot.png
dit xvg_show -f rmsd.xvg -o rmsd_plot.png
dit xvg_show -f potential.xvg -o potential_plot.png
dit xvg_show -f temperature.xvg -o temperature_plot.png
dit xvg_show -f density.xvg -o density_plot.png
dit xvg_show -f pressure.xvg -o pressure_plot.png
افتح مجموعة البيانات التي أنشأناها
5. رسم خريطة مشهد الطاقة GROMACS تم رسم مشهد الطاقة الحرة، مع نصف قطر الدوران والانحراف الجذري المتوسط للانحراف المعياري كمكونين من مكونات تحليل المكونات الرئيسية على التوالي.
gmx_mpi gyrate -s md_0_1.tpr -f md_0_1_noPBC.xtc -o rg.xvg
gmx_mpi rms -s md_0_1.tpr -f md_0_1_noPBC.xtc -o rmsd.xvg
vim rmsd.xvg
#输入以下命令删除以 # 或 @ 开头的行:
:g/^[@#]/d
vim rg.xvg
#输入以下命令删除以 # 或 @ 开头的行:
:g/^[@#]/d
#注意不要有空行,
paste rmsd.xvg rg.xvg > rmsd-rg.xvg
(base) # tail -f rmsd-rg.xvg
9910.0000000 0.0925907 9910 1.37624 1.20925 1.20765 0.931324
9920.0000000 0.0881348 9920 1.38369 1.22248 1.21078 0.932077
9930.0000000 0.0911074 9930 1.39224 1.23799 1.21709 0.928842
9940.0000000 0.0893596 9940 1.38188 1.21942 1.21672 0.922916
9950.0000000 0.0915931 9950 1.37509 1.21939 1.20194 0.922051
9960.0000000 0.0978161 9960 1.38113 1.2262 1.21084 0.919414
9970.0000000 0.0954911 9970 1.37934 1.21241 1.20711 0.937075
9980.0000000 0.0993617 9980 1.38301 1.22353 1.21083 0.92862
9990.0000000 0.1069279 9990 1.37943 1.22317 1.20579 0.924978
10000.0000000 0.1055321 10000 1.37524 1.21648 1.20194 0.92632
^Z
[11]+ Stopped tail -f rmsd-rg.xvg
#查看已经将两个文件整合在一起了,我们只要保留
#从 rmsd-rg.xvg 文件内容可以看到,每一行的数据格式如下:
时间_RMSD(ns) RMSD 时间_Rg(ps) Rg 其他列
(base) tail -f rmsd.xvg
9910.0000000 0.0925907
9920.0000000 0.0881348
9930.0000000 0.0911074
9940.0000000 0.0893596
9950.0000000 0.0915931
9960.0000000 0.0978161
9970.0000000 0.0954911
9980.0000000 0.0993617
9990.0000000 0.1069279
10000.0000000 0.1055321
^Z
[12]+ Stopped tail -f rmsd.xvg
#整理数据为
时间 (ns) RMSD Rg
(base) python
Python 3.8.15 (default, Nov 24 2022, 15:19:38)
[GCC 11.2.0] :: Anaconda, Inc. on linux
Type "help", "copyright", "credits" or "license" for more information.
# 加载数据
>>> import pandas as pd
>>> data = pd.read_csv("rmsd-rg.xvg", delim_whitespace=True, header=None, comment="#")
# 保留需要的列:第 1 列 (RMSD 时间) 、第 2 列 (RMSD) 、第 4 列 (Rg)
>>> cleaned_data = data[[0, 1, 3]]
# 将列名修改为更直观的名称
>>> cleaned_data.columns = ["Time (ps)", "RMSD", "Rg"]
>>> cleaned_data.to_csv("rmsd-rg-cleaned.xvg", sep="\t", index=False, header=False)
>>> exit()
#整理成功
tail -f rmsd-rg-cleaned.xvg
9910.0 0.0925907 1.37624
9920.0 0.0881348 1.38369
9930.0 0.0911074 1.39224
9940.0 0.0893596 1.38188
9950.0 0.0915931 1.37509
9960.0 0.0978161 1.38113
9970.0 0.0954911 1.37934
9980.0 0.0993617 1.38301
9990.0 0.1069279 1.37943
10000.0 0.1055321 1.37524
gmx_mpi sham -tsham 300 -nlevels 100 -f rmsd-rg-cleaned.xvg -ls gibbs.xpm -g gibbs.log -lsh enthalpy.xpm -lss entropy.xpm
dit xpm_show -f gibbs.xpm -o gibbs_2d.png
dit xpm_show -f gibbs.xpm -m 3d -o gibbs_3d.png
#tsham : 设定温度
#-nlevels: 设定 FEL 的层次数量
تعتبر المناظر الطبيعية للطاقة الحرة (FES) أداة مهمة للغاية في المحاكاة الجزيئية، والتي تستخدم بشكل أساسي لوصف توزيع الطاقة الحرة للأنظمة الجزيئية عند إحداثيات محددة. وتتمثل وظيفتها الأساسية في الكشف عن الاستقرار الديناميكي الحراري والعملية الحركية للنظام، وهو ما يكون له أهمية كبيرة في مجالات البحث العلمي التالية:
- دراسات الحالة المستقرة وحالة الانتقال:الكشف عن التكوين المستقر (نقطة الحد الأدنى للطاقة الحرة) ومسار التحويل التكويني (حاجز الطاقة الحرة) للنظام، والذي يستخدم لتحليل طي البروتين والتفاعلات الكيميائية وعمليات التعرف الجزيئي.
- المسارات الحركية والاستقرار الديناميكي الحراري:إن تحديد الفرق في الطاقة الحرة بين الحالات المختلفة يساعد على فهم القوة الدافعة للطاقة في التفاعلات الجزيئية والتحولات التكوينية.
- تصميم الأدوية وأبحاث التحفيز:التنبؤ بمسارات ربط الربيطة بالمستقبلات، والحواجز الطاقية، ومعدلات التفاعل، وتوفير التوجيه النظري لتحليل الآلية الجزيئية والتحسين الوظيفي.
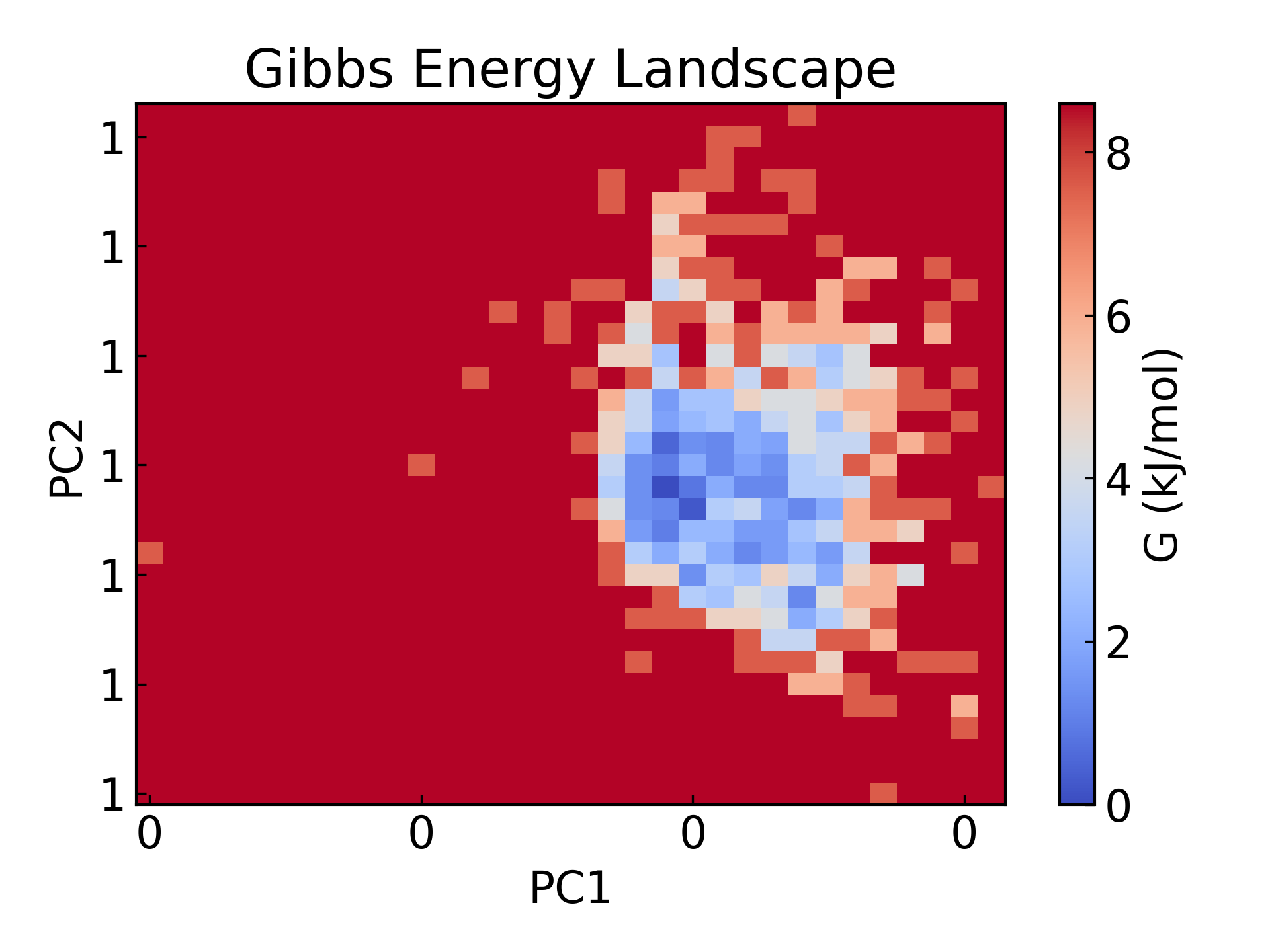
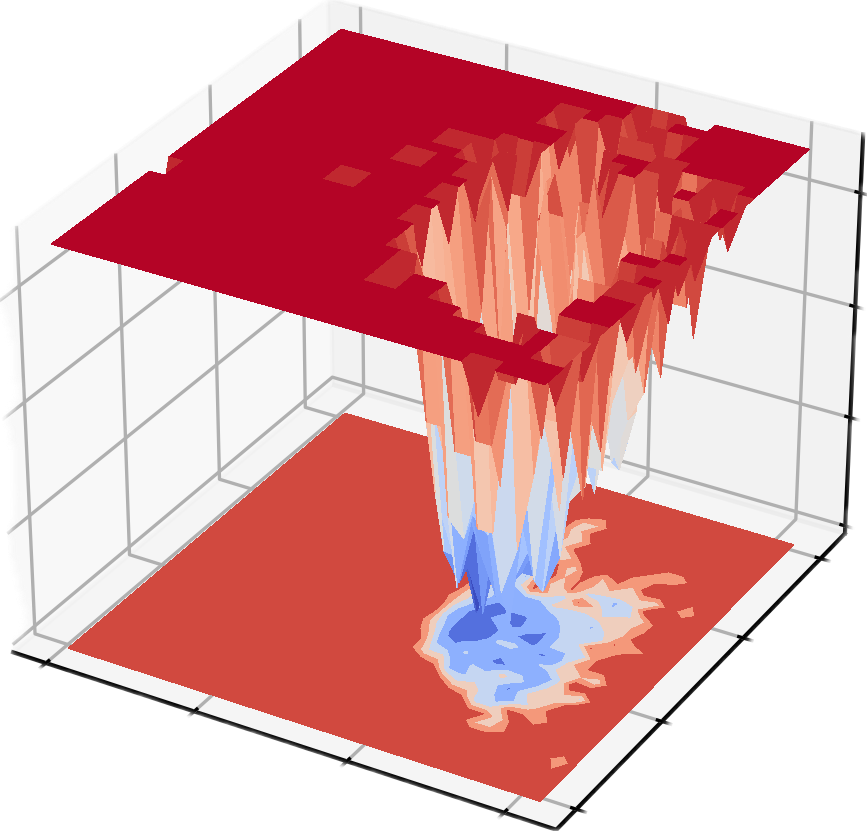
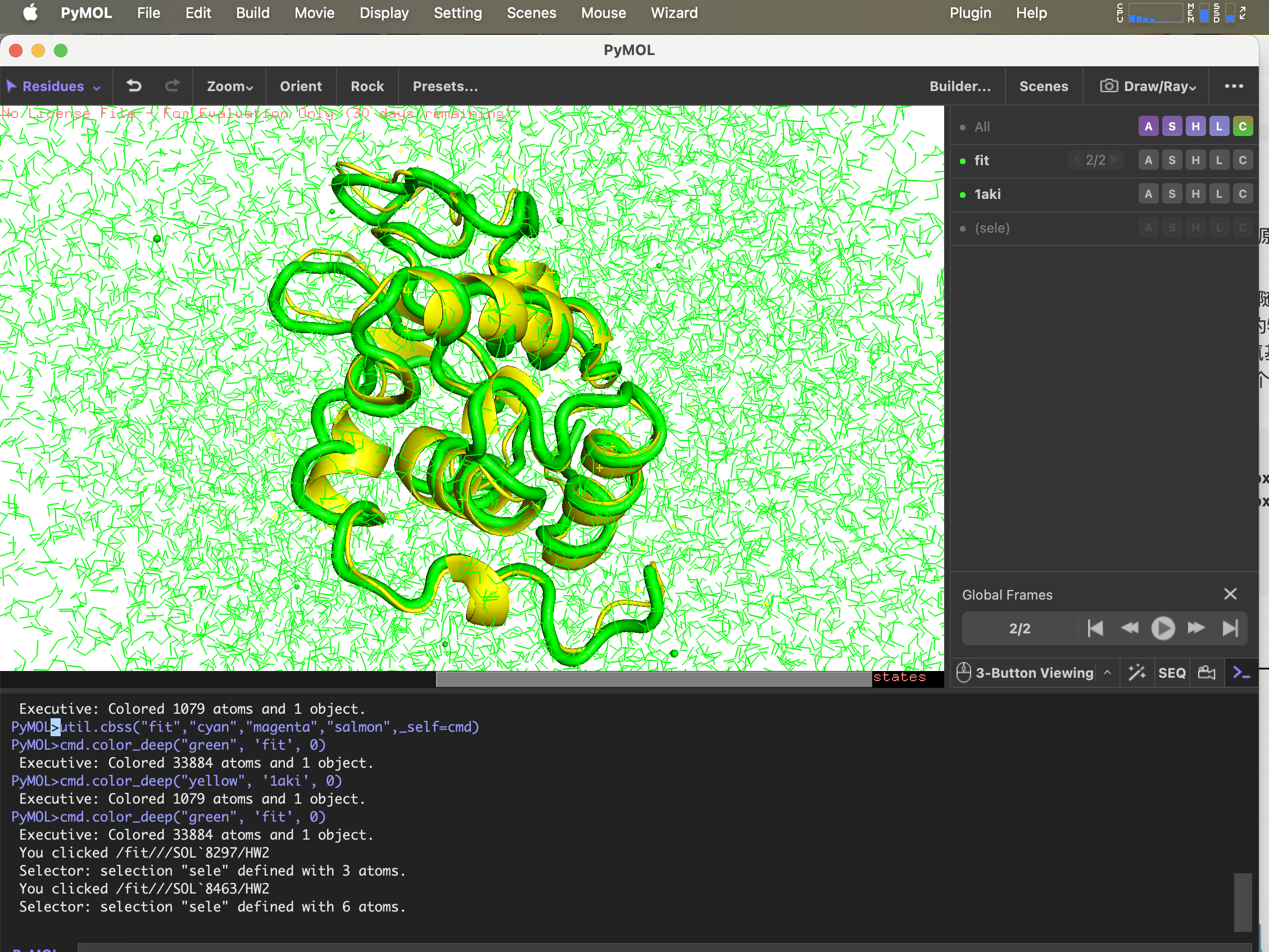
6.g_confrms مقارنة الاختلافات التكوينية
لمقارنة الهيكل بعد المحاكاة بالهيكل الموجود في ملف PDB الأصلي،fit.pdb ملف يحتوي على بنيتين جزيئيتين، حدد كليهما 4 (Backbone)
gmx_mpi confrms -f1 1AKI_clean.pdb -f2 md_0_1.gro -o fit.pdb
تم فتح هيكل قاعدة بيانات البروتين باستخدام Pymol
7.g_rmsf احسب متوسط التقلب التربيعي الجذري (RMSF) والبنية المتوسطة ضمن الـ 500 بيكو ثانية القادمة.g_rmsf النتيجة هي منحنى يتغير مع العدد الذري. يختار 1 Protein
يمكن استنتاج تقلبات بقايا الأحماض الأمينية باستخدام معلمة RMSF، والتي تشرح الانحراف المتوسط لكل بقايا/ذرة من الأحماض الأمينية عن موضع مرجعي بمرور الوقت. بل يمكن القول أنه يقوم بتحليل أجزاء محددة من بنية البروتين التي تختلف عن بنيته المتوسطة. تشير الأحماض الأمينية أو مجموعات الأحماض الأمينية ذات قيم RMSF العالية إلى أن المركب يتمتع بمرونة أكبر، في حين تشير الأحماض الأمينية ذات قيم RMSF المنخفضة إلى أن المركب يتمتع بمرونة أقل. التقلبات المتكررة تؤدي إلى استقرار أسوأ. قيمة RMSF هي معلمة ديناميكية تستخدم لقياس متوسط مرونة العمود الفقري لكل موضع بقايا [19].
gmx_mpi rmsf -s md_0_1.tpr -f md_0_1_noPBC.xtc -b 500 -o fws-rmsf.xvg -ox fws-avg.pdb
gmx_mpi rmsf -s md_0_1.tpr -f md_0_1_noPBC.xtc -b 500 -o fws-rmsf.xvg -ox fws-avg.pdb -res
dit xvg_show -f fws-rmsf.xvg -o rmsf_res.png
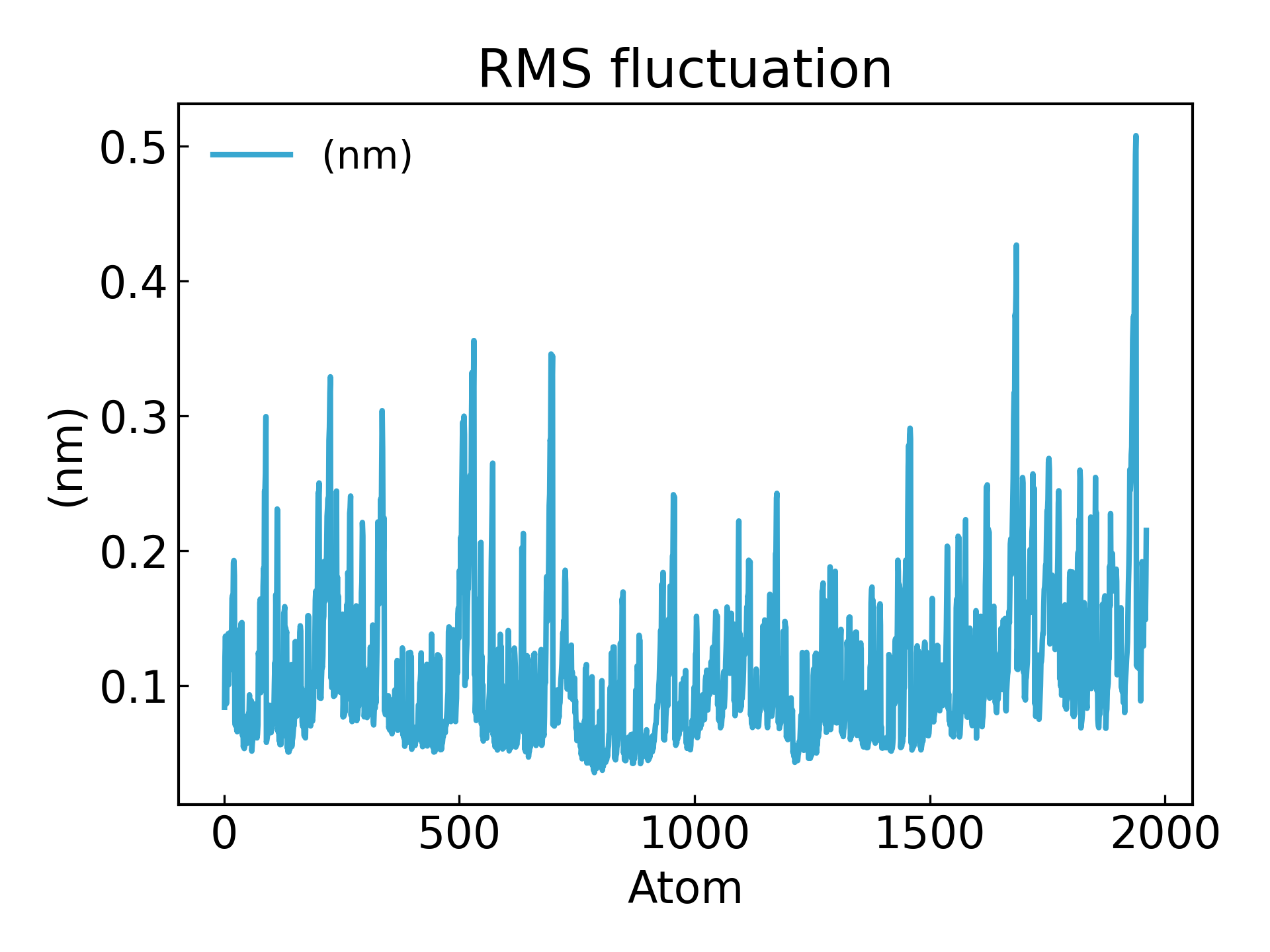
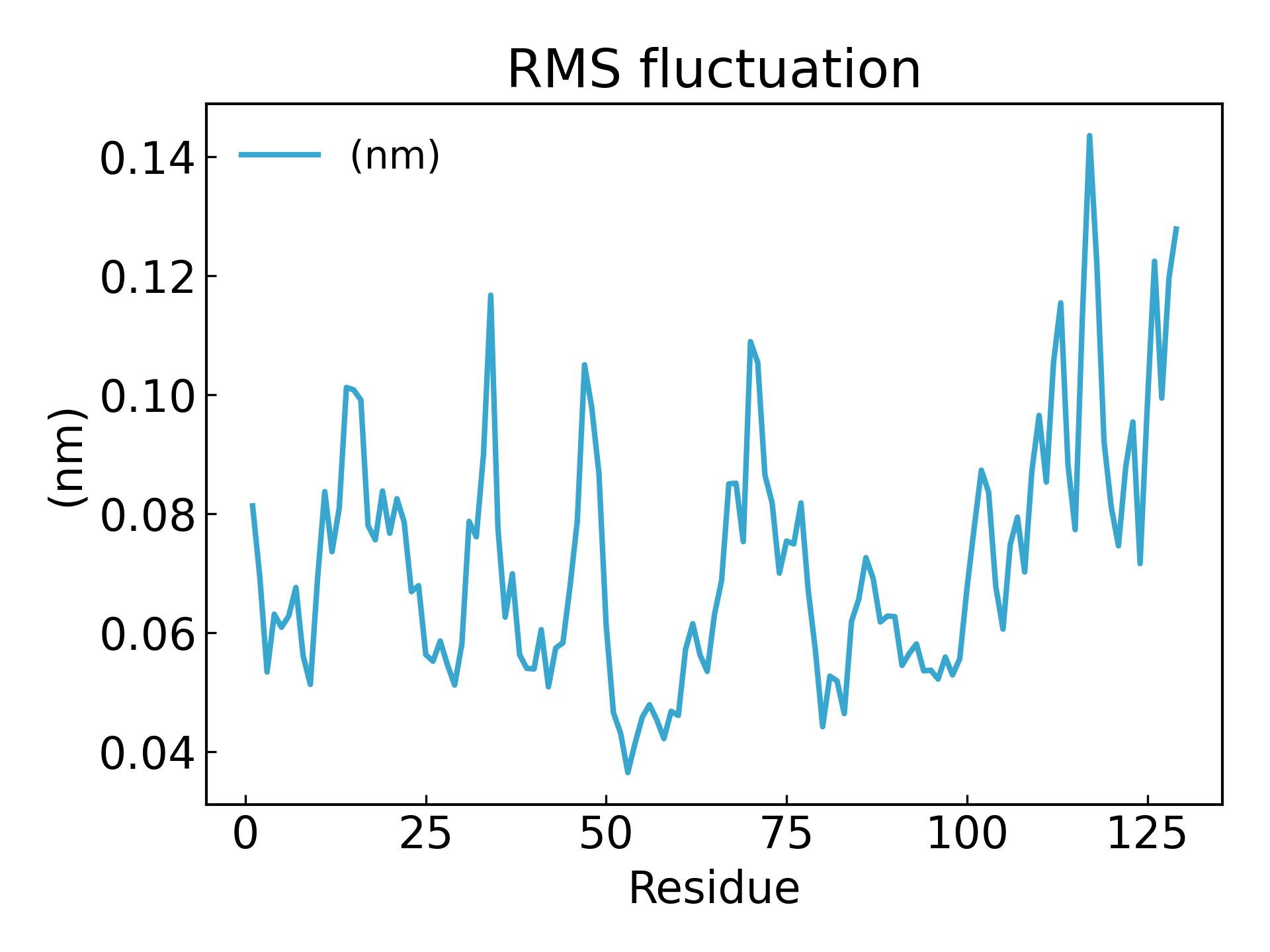
# 最后可以放到 pymol 里面看轨迹动画,点击播放即可
gmx_mpi trjconv -s md_0_1.tpr -f md_0_1_noPBC.xtc -o trajectory.pdb -skip 10
مرجع
بناء الذكاء الاصطناعي بالذكاء الاصطناعي
من الفكرة إلى الإطلاق — سرّع تطوير الذكاء الاصطناعي الخاص بك مع المساعدة البرمجية المجانية بالذكاء الاصطناعي، وبيئة جاهزة للاستخدام، وأفضل أسعار لوحدات معالجة الرسومات.