Command Palette
Search for a command to run...
من خلال معالجة معلومات السلسلة الرئيسية والسلسلة الجانبية للبروتين في وقت واحد، تمكن ستانفورد وآخرون من تحقيق نمذجة بنية الذرة الكاملة استنادًا إلى الشبكة العصبية لتمرير الرسائل
يشير تكوين السلسلة الجانبية للبروتين إلى الترتيب المكاني المحدد للسلاسل الجانبية لبقايا الأحماض الأمينية في البروتينات في الفضاء ثلاثي الأبعاد. تُساعد دراسة تكوين السلسلة الجانبية للبروتين على فهم العلاقة بين بنية البروتين ووظيفته، ولها تطبيقات واسعة في هندسة البروتينات وتصميم الأدوية وغيرها من المجالات. مع ذلك، تُركز أساليب تصميم تسلسلات البروتين الحالية القائمة على التعلم العميق بشكل أساسي على تصميم تسلسلات بروتين السلسلة الرئيسية الثابتة، ومعظمها لا يستطيع نمذجة تكوين السلسلة الجانبية للبروتين أثناء توليد التسلسلات.يتم استنتاج التفاعلات الرئيسية للسلسلة الجانبية فقط بناءً على هندسة السلسلة الرئيسية وعلامات تسلسل الأحماض الأمينية المعروفة، مع تجاهل دور تكوين السلسلة الجانبية للبروتين في البروتينات.
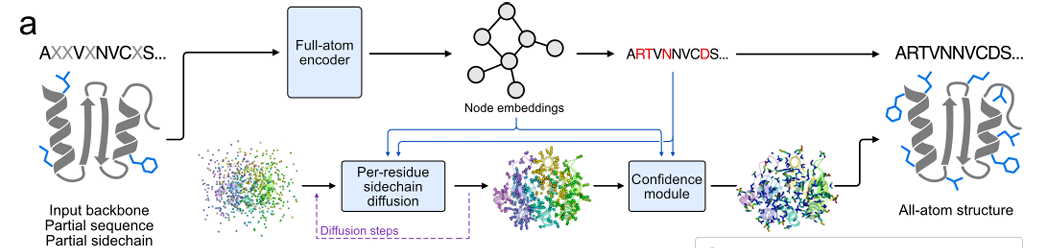
ولسد هذه الفجوة، قام فريق من جامعة ستانفورد ومعهد آرك في بالو ألتو، كاليفورنيا،لقد اقترحنا معًا طريقة جديدة لتصميم تسلسل البروتين، FAMPNN (Full-Atom MPNN)، والتي يمكنها نمذجة هوية التسلسل وبنية السلسلة الجانبية لكل بقايا الأحماض الأمينية بشكل صريح.يستخدم النموذج بنيةً لتمرير الرسائل تعتمد على الشبكات العصبية البيانية (GNNs)، مُدمجةً مع وحدات مُحسّنة من الشبكات العصبية لتمرير الرسائل (MPNN) ووحدات مُستقبلات المتجهات الهندسية (GVP) للترميز الذري الكامل، ويمكنه معالجة معلومات السلسلة الرئيسية والسلسلة الجانبية للبروتين في آنٍ واحد. وقد أظهرت الدراسات أن FAMPNN يُمكن أن يُحسّن بشكل كبير جودة تصميم تسلسل البروتين ودقة التنبؤات التجريبية من خلال النمذجة الصريحة للهياكل الذرية الكاملة.
تم اختيار نتيجة البحث، بعنوان "تكييف السلسلة الجانبية والنمذجة لتصميم تسلسل البروتين الكامل للذرة باستخدام FAMPNN"، لـ ICML 2025.
أبرز الأبحاث:
* نقدم طريقة تجمع بين أهداف فقدان الانتروبيا المتقاطعة والانتشار لنمذجة توزيع كل من هوية التسلسل المنفصل للبقايا وبنية السلسلة الجانبية المستمرة لكل علامة.
* نقوم بتنفيذ طريقة أخذ عينات تكرارية خفيفة الوزن لتوليد عينات من توزيع مشترك واستخدام طبقات MPNN وGVP المحسنة للترميز الذري الكامل
* أظهرت الأبحاث أن FAMPNN يمكن أن يحسن بشكل فعال دقة تصميم التسلسل والتنبؤ بملاءمة البروتين التجريبية من خلال النمذجة الصريحة لجميع هياكل الذرات

عنوان الورقة:
مزيد من أوراق البحث الرائدة في مجال الذكاء الاصطناعي:
https://go.hyper.ai/owxf6
مجموعات البيانات: تعمل مجموعات البيانات المتنوعة على تحسين تدريب النموذج وتقييمه
ولضمان فعالية وموثوقية النموذج، استخدم فريق البحث مجموعات بيانات متعددة ومعقدة للتدريب والتقييم:
استخدمت الدراسة بشكل أساسي مجموعة بيانات S40 من CATH 4.2.تتكون مجموعة البيانات من مجموعة مختارة من المجالات المستخرجة من بنك بيانات البروتين (PDB)، مع إزالة المجالات المكررة ذات التشابه الذي يتجاوز 40%، وتقسيمها إلى مجموعات تدريب وتحقق واختبار بنسبة 8:1:1.
تم بناء مجموعة بيانات PDB بناءً على قاعدة بيانات PDB بأكملها وتتضمن الهياكل المنشورة اعتبارًا من 30 سبتمبر 2021. قام الباحثون بتجميع البروتينات وفقًا لتشابه تسلسل 40% على مستوى سلسلة البروتين، وأعطوا الأولوية لأمثلة البروتين متعدد السلاسل لتدريب النماذج لتعلم تصميم البروتينات متعددة السلاسل.
تُستخدم مجموعات البيانات CASP13 و14 و15 بشكل أساسي لتقييم أداء النموذج في تعبئة السلسلة الجانبية.استخدم فريق البحث البحث عن طريق اقتراح MMseqs2 لإزالة جميع التسلسلات ذات التشابه الأكبر من 40% مع مجموعات البيانات CASP13 و14 و15 من مجموعات البيانات التدريبية والتحققية، ثم قاموا بقياس أداء تعبئة السلسلة الجانبية من خلال متوسط انحراف الجذر التربيعي المتوسط (RMSD) بين السلاسل الجانبية المتوقعة والسلاسل الجانبية الحقيقية.
تم استخدام مجموعة بيانات SKEMPlv2 لتقييم قدرة النموذج التنبؤية على ربط البروتين بالبروتين.تجمع مجموعة البيانات تقارب الارتباط المقاس تجريبياً لآلاف المتغيرات التسلسلية في مئات من التفاعلات بين البروتينات، وبعد المعالجة، تتكون مجموعة البيانات النهائية من 6649 نقطة بيانات.
تم استخدام مجموعات البيانات S669 وMegascale وFireProtDB لتقييم قدرة النموذج على التنبؤ بالخطأ الصفري لاستقرار البروتين.تحتوي مجموعات البيانات هذه على قياسات تجريبية لتغيرات استقرار مجموعة متنوعة من البروتينات الطبيعية (△△G)، وهي مجموعات بيانات مرجعية تُستخدم على نطاق واسع للتنبؤ بالاستقرار. مجموعة بيانات Megascale هي نسخة مختارة من مجموعة البيانات، خالية من التكرار. دمج فريق البحث مجموعة التدريب ومجموعة التحقق ومجموعة الاختبار في مجموعة بيانات واحدة، وحصل في النهاية على مجموعة بيانات تحتوي على 272,712 نقطة بيانات تجريبية تشمل 298 بروتينًا مختلفًا. تحتوي مجموعة بيانات FireProtDB على تغيرات في الطاقة الحرة لـ 3,438 طفرة مفردة لـ 100 بروتين فريد، استُخدم منها 3,420 مثالًا بعد المعالجة. تحتوي مجموعة بيانات S669 على قياسات تجريبية لـ 669 طفرة مفردة لـ 94 بروتينًا، وتم استبعاد 4 متغيرات من مجموعة البيانات بسبب وجود أحماض أمينية غير قياسية.
تم استخدام مجموعات البيانات CR9114 وCR6261 وG6 لتقييم أداء النموذج في التنبؤ بتقارب ارتباط الأجسام المضادة بالمستضد.من بينها، تحتوي مجموعة بيانات CR9114 على جميع التركيبات الممكنة لـ 16 استبدالًا للأحماض الأمينية. وتحتوي مجموعة بيانات CR6261 على جميع التركيبات الممكنة لـ 11 استبدالًا للأحماض الأمينية، بإجمالي 65,536 و2,048 تسلسلًا على التوالي. وتحتوي مجموعة بيانات G6 على إجمالي 4,275 نقطة بيانات مرتبطة بـ VEGF-A.
أداة ذكية لفهم تسلسل البروتين وبنية السلسلة الجانبية في نفس الوقت
الهدف الأساسي من هذه الدراسة هو تمكين النموذج من تعلم تسلسل البروتين وتكوين السلسلة الجانبية في آنٍ واحد. ولتحقيق ذلك، استخدم فريق البحث نمذجة اللغة المقنعة لتدريب FAMPNN بناءً على اتساق التسلسل.يتم إجراء التدريب بطريقة شاملة، من خلال الجمع بين فقدان الإنتروبيا المتقاطعة التصنيفية (للتنبؤ بالتسلسل) وفقدان الانتشار (للتنبؤ بتكوين السلسلة الجانبية).يتيح هذا للنموذج استعادة التسلسل المقنع وتكوينات السلسلة الجانبية المقابلة له في وقت واحد استنادًا إلى التسلسلات المعروفة جزئيًا وإحداثيات السلسلة الجانبية.
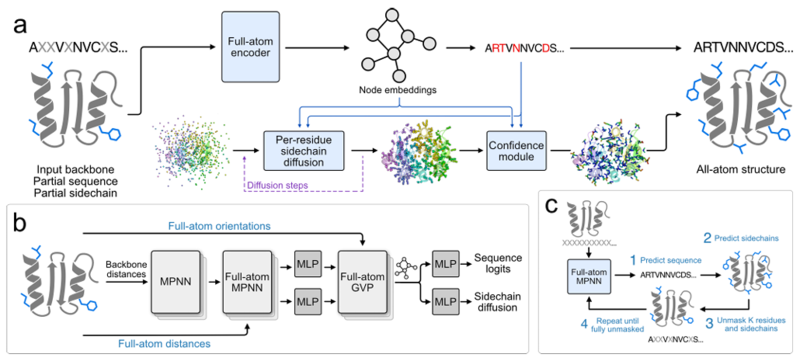
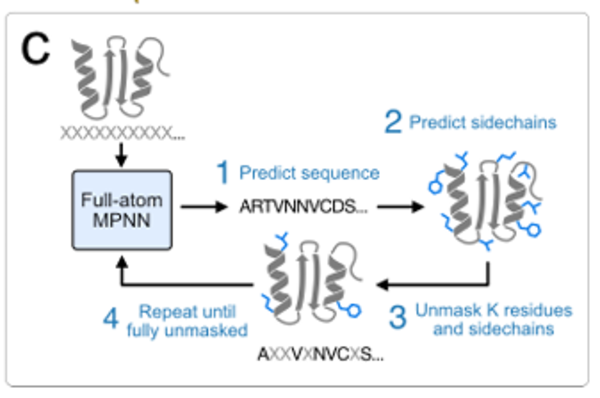
فيما يتعلق بأخذ العينات، تُقدّم استراتيجية تكرارية لأخذ العينات، تُشبه استراتيجية MaskGIT، بدءًا من حالة إخفاء التسلسل والسلسلة الجانبية بالكامل، ثم التنبؤ تدريجيًا بإزالة قناع بعض التسلسلات ورموز السلسلة الجانبية، حتى يتم الحصول على بنية التسلسل والسلسلة الجانبية كاملةً. كما هو موضح في الشكل أدناه:
في التصميم المحدد، يتم تمثيل إحداثيات السلسلة الجانبية بتنسيق atom37.كل بقايا عبارة عن مصفوفة ثابتة الحجم، أبعادها 37 × 3، تحتوي على 37 ذرة، بما في ذلك إحداثيات ثلاثية الأبعاد لأربع ذرات سلسلة رئيسية (N، Cα، C، وO) و33 ذرة سلسلة جانبية. بالنسبة للسلاسل الجانبية التي لا يوجد فيها نوع ذرات محدد، تُستخدم ذرات شبحية (مُحددة بموضع Cα في البقايا) لتمثيلها. تحل هذه الطريقة مشكلة اختلاف عدد ذرات السلسلة الجانبية باختلاف الأحماض الأمينية.
باعتباره جوهر استخراج الميزات، يستخدم مشفر الذرات بالكامل شبكة عصبية بيانية ذات بنية هجينة MPNN-GVP للترميز.تتكون البنية من ثلاثة مكونات رئيسية: مُشفِّر السلسلة الرئيسية الثابت، ومُشفِّر الذرات الكامل الثابت، ومُشفِّر الذرات الكامل المتساوي التغير. بُني المكونان الأولان على بنية ProteinMPNN. مُشفِّر السلسلة الرئيسية الثابت هو نفسه مُشفِّر MPNN، الذي يُشفِّر بنية السلسلة الرئيسية فقط؛ بينما يحل مُشفِّر الذرات الكامل الثابت محل مُفكِّك تشفير MPNN، وهو نفسه مُشفِّر MPNN الخاص بمُشفِّر السلسلة الرئيسية، ولكن التوصيف مُمتد ليشمل جميع الذرات. يستخدم المكون الأخير نموذج GVP مُحسَّنًا لتمكين النموذج من التفكير في التوجهات بين الذرات ذات القيم المتجهة، بالإضافة إلى المسافات بين الذرات ذات القيم القياسية المُشفَّرة سابقًا.
من حيث توليد إحداثيات السلسلة الجانبية، اعتمد فريق البحث طريقة الانتشار الإقليدي لكل رمز.يعتمد جوهر هذه الطريقة على استخدام نموذج الانتشار الإقليدي (EDM) لحل مشكلة توليد قيم متصلة لإحداثيات ذرات السلسلة الجانبية. الهدف هو توليد بنية سلسلة جانبية تتوافق مع بنية السلسلة الرئيسية والترتيب المكاني للأحماض الأمينية المحيطة. أثناء التدريب، تُضاف ضوضاء عشوائية أولًا إلى إحداثيات السلسلة الجانبية الحقيقية، ثم يُتاح للنموذج إزالة الضوضاء واستعادة الإحداثيات الحقيقية بناءً على مستوى الضوضاء والمعلومات المعروفة. أثناء الاستدلال، بدءًا من إحداثيات الضوضاء العشوائية، يُزيل النموذج الضوضاء تدريجيًا ويُولّد إحداثيات سلسلة جانبية قريبة من إحداثياتها الحقيقية.
في الوقت نفسه، من أجل تجنب تأثير الدوران الشامل وترجمة البروتين على توليد السلسلة الجانبية، يتم تحويل إحداثيات ذرات السلسلة الجانبية إلى نظام إحداثيات محلي يعتمد على ذرات السلسلة الرئيسية أثناء التدريب، ثم يتم تحويلها مرة أخرى إلى نظام الإحداثيات العالمي بعد التوليد.يتضمن إدخال نموذج الانتشار الميزات المستخرجة بواسطة مشفر الذرات بالكامل، وهوية التسلسل المتوقعة ومستوى الضوضاء الحالي.يتم أيضًا استخدام إحداثيات السلسلة الجانبية المولدة في دالة الخسارة المشتركة لتوجيه تدريب النموذج، كما هو موضح في الشكل أدناه.
لتقليل خطأ التنبؤ بالنموذج وتحسين دقته، صمم فريق البحث أيضًا وحدة ثقة للتنبؤ بخطأ تعبئة السلسلة الجانبية (خطأ السلسلة الجانبية المتوقع، pSCE). وتحديدًا،تقوم هذه الوحدة بتقسيم الخطأ الفعلي لذرات السلسلة الجانبية (المسافة بين الإحداثيات المولدة والإحداثيات الحقيقية) إلى 33 فاصلًا.تم تدريب النموذج باستخدام فقدان الإنتروبيا المتقاطعة الفئوية، مما يسمح له بالتنبؤ بفاصل كل خطأ ذري بناءً على المعلومات في عملية التوليد، ومن ثم الحصول على تقدير الخطأ النهائي pSCE من خلال توقع احتمالية الفاصل. تتضمن مدخلات هذه الوحدة خصائص مُشفِّر الذرات بالكامل، والتسلسل المُولَّد، وإحداثيات السلسلة الجانبية، ومستوى الضوضاء في عملية الانتشار. يعكس مُخرَج pSCE بدقة دقة تعبئة السلسلة الجانبية، مما يُساعد في فحص نتائج التصميم عالية الجودة وتعزيز قابلية تفسير النموذج، وبالتالي تحسين جودة رابط تقييم توليد بنية السلسلة الجانبية.
النتائج التجريبية: الأداء أفضل بكثير من النموذج الذي يعتمد فقط على السلسلة الرئيسية
للتحقق من الأداء وتقييم النموذج بدقة، أجرى فريق البحث أولًا تجارب استعادة التسلسل وتقييم الاتساق الذاتي، وقارن أداء FAMPNN بأساليب أخرى. تظهر عناصر المقارنة المحددة في الشكل أدناه.
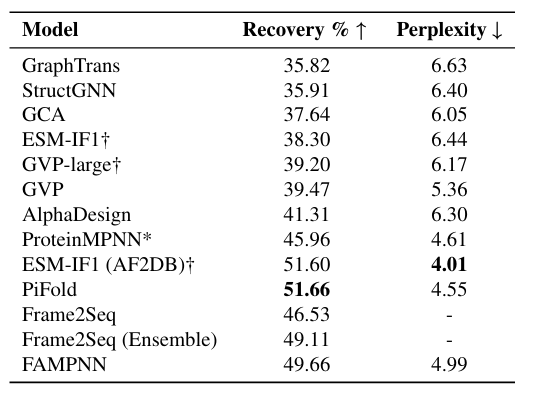
تظهر التجارب أنيتفوق FAMPNN على الطرق الحديثة المتطورة من حيث دقة استرداد التسلسل بخطوة واحدة، حيث يصل إلى 49.66%.بالمقارنة، فإن ProteinMPNN هو 45.96% فقط وGVP هو 39.47% فقط.
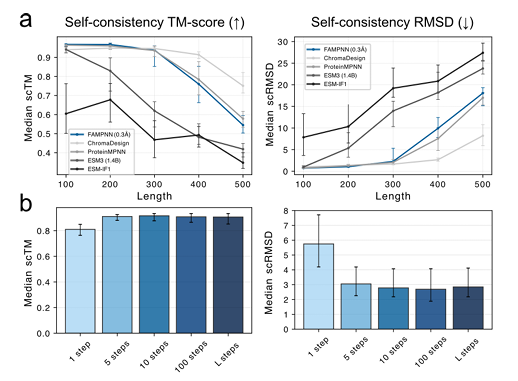
في تقييم الاتساق الذاتي على السلسلة الرئيسية الجديدة المولدة بناءً على انتشار الترددات الراديوية،FAMPNN (0.3Å) قابل للمقارنة مع ProteinMPNN من حيث مقاييس scTM (التشابه البنيوي) وscRMSD (الانحراف الجذري المتوسط التربيعي).ويمكن لعشر خطوات من أخذ العينات التكرارية تحقيق درجة عالية من الاتساق الذاتي، وهي أكثر كفاءة من طريقة الانحدار الذاتي الكامل. كما هو موضح في الشكل التالي:
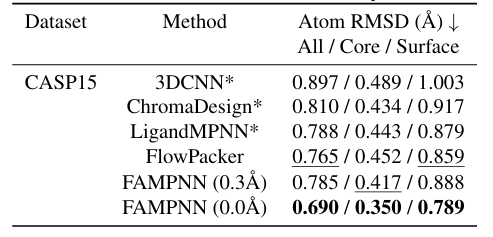
وفيما يتعلق بتعبئة السلسلة الجانبية، قارن الباحثون النموذج المقترح مع طرق أخرى على مجموعات البيانات CASP13 و14 و15.تظهر التجارب أنه في تقييم البنية البلورية لمجموعة اختبار CASP15، فإن RMSD الذري (الكل/النواة/السطح) لـ FAMPNN (0.0Å) هو 0.690/0.350/0.789Å، وهو أفضل من الطرق الأخرى.وله ارتباط قوي مع خطأ كل ذرة وخطأ كل بقايا، بمعاملي ارتباط سبيرمان ٠.٨٤٣ و٠.٧٨٠ على التوالي. كما هو موضح في الشكل أدناه:
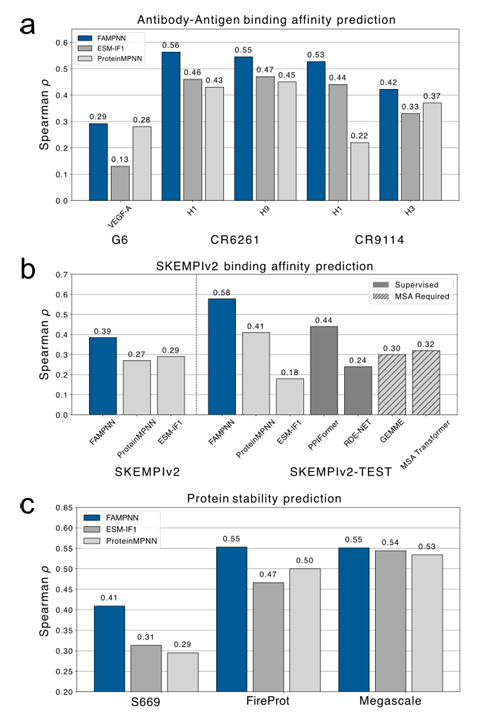
في تقييم لياقة البروتين في ظل ظروف جميع الذرات،في مجموعة بيانات SKEMPlv2، تفوق FAMPNN بشكل ملحوظ على النموذج غير الخاضع للإشراف، بل وتفوق عليه في مجموعة الاختبار الفرعية، مُظهرًا قدرة تعميم قوية في التنبؤ بالعينة الصفرية. في مجموعات بيانات الاستقرار الثلاث S669 وMegascale وFireProtDB، كان أداء FAMPNN أفضل بقليل من ProteinMPNN وESM-IF؛ ففي التنبؤ بألفة ارتباط الأجسام المضادة بالمستضد، تفوق FAMPNN دائمًا على أحدث الطرق غير الخاضعة للإشراف، ProteinMPNN وESM-IF1، مما يُثبت فائدة FAMPNN في استقرار البروتين وتعزيز تفاعلاته. كما هو موضح في الشكل التالي:
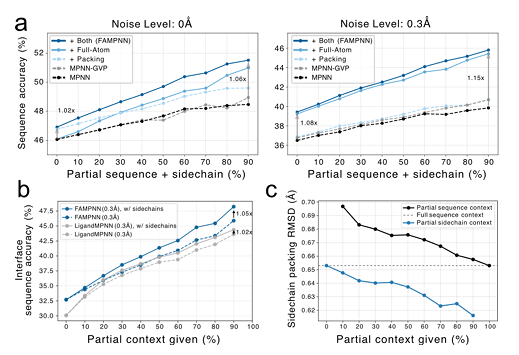
في التجارب التي تهدف إلى تقييم ما إذا كان النمذجة الذرية الكاملة قادرة على تحسين أداء تصميم التسلسل،توصلت الدراسة إلى أن إضافة أهداف تعبئة السلسلة الجانبية وإعدادات حالة الذرة الكاملة يمكن أن يحسن دقة التسلسل.علاوة على ذلك، سيتحسن أداء كلٍّ من FAMPNN والنموذج الأساسي مع إدخال المزيد من المعلومات الهيكلية. عند واجهة البروتين-البروتين، تُعد نمذجة تفاعلات السلسلة الجانبية أكثر أهمية، كما أن توفير سياق جزئي للسلسلة الجانبية بالتزامن مع التسلسل يُحسّن الدقة بشكل ملحوظ مقارنةً بتوفير سياق جزئي للتسلسل فقط.
بالإضافة إلى ذلك، مقارنةً بـ LigandMPNN، يُمكن لـ FAMPNN الاستفادة بشكل أكثر فعالية من سياق السلسلة الجانبية، وإجراء عملية تعبئة السلسلة الجانبية بناءً على أعداد مختلفة من سياقات التسلسل الجزئي أو تكوين السلسلة الجانبية. كلما زاد عدد السياقات، زادت دقة التعبئة. كما هو موضح في الشكل ج أدناه:
باختصار، توضح التجارب المذكورة أعلاه أن FAMPNN يتمتع بمزايا كبيرة في التنبؤ بلياقة البروتين مقارنة بالنماذج التي تعتمد فقط على العمود الفقري.
بفضل الذكاء الاصطناعي، تزدهر الأوساط الأكاديمية في مجال نمذجة السلسلة الجانبية
كما ذُكر سابقًا، يُعدّ تكوين السلسلة الجانبية أساسيًا لوظيفة البروتين. ومع ذلك، بعد تحديد السلسلة الرئيسية للبروتين، لا تزال هناك العديد من التكوينات المحتملة للسلسلة الجانبية، مما يجعل نمذجة وبحث تكوينات السلسلة الجانبية مشكلةً صعبةً يجب التغلب عليها. بالإضافة إلى هذه الدراسة، تُسهم العديد من مؤسسات البحث الأكاديمي حول العالم في دفع أبحاث نمذجة السلسلة الجانبية قدمًا، من خلال تقنيات التعلم العميق المتطورة والمعرفة البيولوجية.
اقترح فريق من جامعة فودان في الصين طريقة نمذجة السلسلة الجانبية المكونة من مرحلتين والتي تسمى OPUS-Rota5.تستخدم هذه الطريقة شبكة 3D-Unet المحسنة لالتقاط ميزات البيئة المحلية.تم تضمين معلومات الربيطة لكل بقايا، ثم استخدام وحدة RotaFormer لتجميع أنواع مختلفة من الخصائص. أظهرت التقييمات على مجموعات اختبار، بما في ذلك CAMEO وCASP15، أن OPUS-Rota5 يتفوق بشكل ملحوظ على بعض طرق نمذجة السلسلة الجانبية الرائدة الأخرى. نُشر البحث ذو الصلة على ScienceDirect بعنوان "OPUS-Rota5: طريقة نمذجة سلسلة جانبية عالية الدقة للبروتين باستخدام 3D-Unet وRotaFormer".
عنوان الورقة:
https://www.sciencedirect.com/science/article/pii/S0969212624001266
واقترح فريق من جامعة بكين طريقة أخرى تسمى GeoPacker.تجمع هذه الطريقة بين التعلم العميق الهندسي وResNet لنمذجة السلاسل الجانبية للبروتين. يُمثل GeoPacker التفاعلات الذرية بوضوح بطريقة ثابتة دورانيًا وانتقاليًا لاستخراج معلومات الموقع النسبي. من حيث دقة التنبؤ ببنية السلسلة الجانبية، يتفوق GeoPacker على أحدث الطرق القائمة على دوال الطاقة، ويعمل أسرع بحوالي 10 مرات و700 مرة من طريقتي التعلم العميق DLPacker وOPUS-Rota4، على التوالي، بدقة تنبؤ مماثلة. نُشر البحث ذو الصلة بعنوان "GeoPacker: إطار عمل جديد للتعلم العميق لنمذجة السلسلة الجانبية للبروتين".
عنوان الورقة:
https://onlinelibrary.wiley.com/doi/epdf/10.1002/pro.4484
وفي الوقت نفسه، اقترح الفريق في جامعة تورنتو نموذجًا يسمى FlowPacker.غرضه هو التنبؤ بدقة بالشكل المحدد للسلسلة الجانبية بناءً على تسلسل الأحماض الأمينية المعروفة وبنية السلسلة الرئيسية للبروتين.مقارنةً بالطرق المتقدمة السابقة، يُظهر FlowPacker أداءً أفضل في معظم المؤشرات وأسرع في العمل. على سبيل المثال، يتميز بمزايا في خطأ التنبؤ بالزاوية، وتقارب الزاوية المتوقعة مع القيمة الحقيقية، وانحراف موضع الذرات، وغيرها. نُشر البحث ذو الصلة تحت عنوان "FlowPacker: تعبئة السلسلة الجانبية للبروتين مع مطابقة التدفق الالتوائي".
عنوان الورقة:
بشكل عام، يُعدّ فكّ تشفير تكوينات السلسلة الجانبية أمرًا بالغ الأهمية لتطوير مجال علوم الحياة. ولا شك أن التطور المستمر لتكنولوجيا الذكاء الاصطناعي قد ساهم في تسريع وتيرة التطور في علم الأحياء البنيوي وعلم الأحياء الحاسوبي، كما ساعد مؤسسات البحث المحلية والأجنبية على تحقيق إنجازات أكاديمية مزدهرة. وبمجرد تطبيق هذه الإنجازات، من المختبر إلى التطبيق، ستُحدث حتمًا ثورةً جديدة في مجال علوم الحياة، وستدفع العلوم البيولوجية والعلوم الطبية نحو آفاق جديدة.








