Command Palette
Search for a command to run...
اقترح معهد ماساتشوستس للتكنولوجيا وجامعة هارفارد بشكل مشترك PUPS من خلال دمج نموذج لغة البروتين ونموذج تلوين الصور لتحقيق توطين البروتين في الخلية الواحدة
يشير التوطين الفرعي للبروتين إلى الموقع المحدد للبروتين في بنية الخلية.وهذا ضروري للبروتينات للقيام بوظائفها البيولوجية. على سبيل المثال البسيط، إذا تخيلنا الخلية كمؤسسة ضخمة، حيث تتوافق نواة الخلية والميتوكوندريا والغشاء الخلوي وما إلى ذلك مع أقسام مختلفة مثل مكتب الرئيس وقسم توليد الطاقة والبواب، فعندئذ فقط عندما يدخل البروتين المقابل إلى "القسم" الصحيح يمكنه العمل بشكل طبيعي، وإلا فإنه سوف يسبب أمراضًا معينة مثل السرطان ومرض الزهايمر. لذلك، يمكن القول بأن تحديد موقع البروتينات تحت الخلوية بدقة يعد أحد المهام الأساسية لعلوم الحياة.
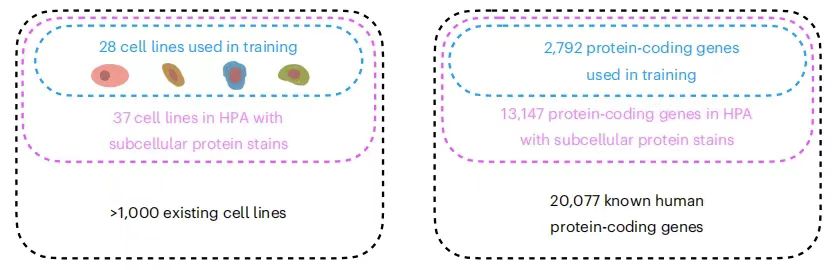
وعلى الرغم من تحليل التوطين المكاني لآلاف البروتينات في خطوط الخلايا المختلفة، فإن عدد تركيبات خطوط الخلايا والبروتينات التي تم قياسها حتى الآن لا يمثل سوى قمة جبل الجليد. على سبيل المثال، أكبر مجموعة بيانات توطين الخلايا الفرعية المتوفرة حاليًا هي:يوفر أطلس البروتين البشري (HPA) توطينًا فرعيًا للبروتينات المشفرة بواسطة 13147 جينًا (بما في ذلك 65% من جينات ترميز البروتين البشري المعروفة).ومع ذلك، تضمنت مجموعة البيانات بأكملها 37 خطًا خلويًا، وتم قياس كل بروتين في ثلاثة منها على الأكثر. وفي الوقت نفسه، تجعل الطرق التجريبية السائدة من الصعب اكتشاف عدد جميع البروتينات في نفس الخلية في نفس الوقت، مما يعوق بشكل خطير التحليل الشامل لشبكات البروتين المعقدة ويزيد من تعقيد التجارب ومخاطر الخطأ.
بالإضافة إلى ذلك، فإن توطين البروتين ليس ثابتًا ويحدث تغيره ليس فقط بين خطوط الخلايا ولكن أيضًا بين الخلايا الفردية في نفس خط الخلايا. تعكس أزواج البروتين وخطوط الخلايا المسجلة في خرائط البيانات الموجودة النتائج في ظل ظروف محددة فقط. لذلك،ولكن حتى النتائج الموجودة يصعب تطبيقها بشكل مباشر، وهناك حاجة إلى مزيد من الاستكشاف لتوطين البروتين على أساس التغيرات البيئية.
ومن المتوقع أن يكون التعلم الآلي واعدًا لحل التناقض بين قيود أساليب تكنولوجيا توطين البروتين تحت الخلوي وتعقيد الأنظمة البيولوجية. لقد حققت النماذج التي تم بناؤها وتطبيقها بنجاح اليوم، مثل النماذج القائمة على تسلسل البروتين والنماذج القائمة على صور الخلايا، أداءً جيدًا في بعض الجوانب، ولكن عيوبها بارزة جدًا أيضًا - حيث تتجاهل النماذج الأولى الاختلافات في التوطين المحدد لأنواع الخلايا، وتفتقر النماذج الثانية إلى القدرة على التعميم لتعزيز دراسة البروتينات غير المعروفة.
وفي ضوء ذلك،اقترح فريق بحثي من معهد ماساتشوستس للتكنولوجيا وجامعة هارفارد إطار عمل للتنبؤ بالتوطين الفرعي للبروتينات غير المعروفة من خلال الجمع بين تسلسلات البروتين وصور الخلايا، وأطلق عليه اسم "تنبؤات التوطين الفرعي للبروتينات غير المرئية" (PUPS). يجمع PUPS بشكل مبتكر بين نموذج لغة البروتين ونموذج طلاء الصور للتنبؤ بموقع البروتين، مما يتيح له دمج قدرات التعميم لتوقعات البروتين غير المعروفة مع التنبؤات الخاصة بنوع الخلية والتي تلتقط التباين الخلوي. وقد أظهرت التجارب أن هذا الإطار يمكنه التنبؤ بدقة بموقع البروتينات في التجارب الجديدة خارج مجموعة بيانات التدريب، ولديه قدرة تعميم ممتازة ودقة عالية، ولديه إمكانات تطبيقية متميزة.
وقد تم نشر نتائج البحث، بعنوان "التنبؤ بتوطين البروتين الفرعي في الخلايا الفردية"، في مجلة Nature Methods.
أبرز الأبحاث:
* تجمع الدراسة المقترحة بشكل مبتكر بين نماذج لغة البروتين ونماذج تقديم الصور، باستخدام تسلسلات البروتين وصور الخلايا للتنبؤ بموقع البروتين، مما يعوض عن أوجه القصور في النماذج الحسابية السابقة
* PUPS قادر على التعميم على البروتينات وخطوط الخلايا غير المعروفة، وبالتالي تقييم التباين في توطين البروتين بين خطوط الخلايا وبين الخلايا الفردية داخل خط الخلية، وتحديد العمليات البيولوجية المرتبطة بالبروتينات ذات التوطين المتغير
* في تجارب جديدة خارج مجموعة بيانات التدريب، أثبت PUPS أيضًا قدرته على التنبؤ بدقة عالية، مع إمكانات تطبيقية متميزة وقيمة طبية

عنوان الورقة:
مجموعات البيانات: بناء نماذج جديرة بالثقة مع البيانات الأكثر شمولاً الممكنة
تأتي مجموعة بيانات تدريب PUPS من أطلس البروتين البشري (HPA).قام فريق البحث بتجميع النسخة السادسة عشر من بيانات HPA في النسخة الثانية والعشرين لجمع أكبر قدر ممكن من بيانات البروتين وضمان شمولية التحليل التجريبي. كما هو موضح في الشكل التالي:
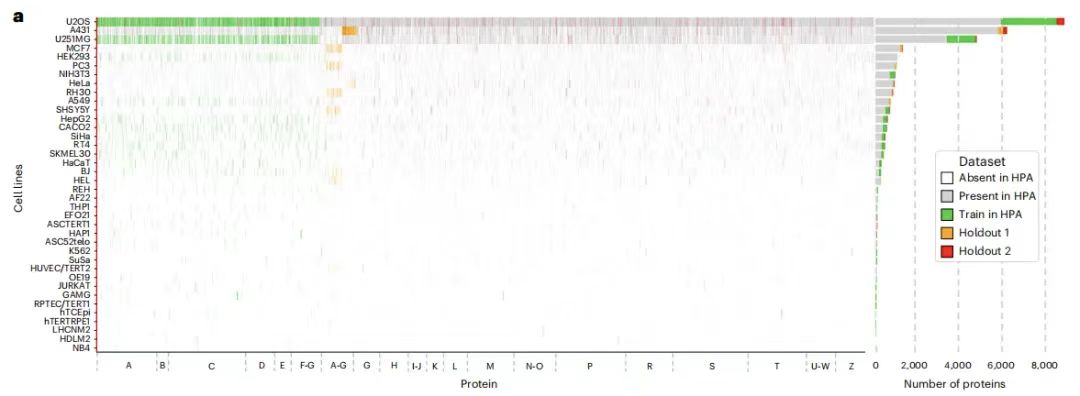
على وجه التحديد، تحتوي مجموعة بيانات التدريب على 340,553 مجموعة خلوية بإجمالي 8,086 متغيرًا بروتينيًا يتوافق مع 2,801 جينًا في 37 سلالة خلوية في HPA، والتي تبدأ أسماؤها بالأحرف AG. بالإضافة إلى ذلك، تتضمن مجموعة بيانات التدريب أيضًا 10 جينات إضافية، بما في ذلك IHO1، وIMPAD1، وINKA1، وISPD، وITPRID1، وKIAA1211L، وKIAA1324، وLRATD1، وSCYL3، وTSPAN6.
تنقسم مجموعة البيانات الممتنعة إلى قسمين:جزء واحد هو مجموعة البيانات المحجوزة 1،تحتوي على 36,552 خلية، مع 9,472 متغيرًا بروتينيًا يتوافق مع 3,312 جينًا (بما في ذلك 2,801 في مجموعة التدريب)، والتي تبدأ أسماؤها أيضًا بحرف AG ولكنها تأتي من خطوط خلوية مختلفة وليس لها تداخل مع مجموعة التدريب. وفي الوقت نفسه، تم تقسيم مجموعة البيانات المحتجزة 1 إلى قسمين واستخدامها كمجموعة تقييم ومجموعة اختبار، تحتوي على 11050 و25502 خلية على التوالي؛تحتوي مجموعة البيانات المحفوظة 2 على 24007 خلية، تتوافق مع 515 جينًا.يبدأ اسمه بجميع حروف الأبجدية، ويغطي الألف إلى الياء. هناك 556 متغيرًا بروتينيًا في المجموع، والتي تأتي من عائلات جينية جديدة لا تظهر في مجموعة التدريب ومجموعة البيانات المحجوزة1 ويمكن استخدامها لاختبار قدرة النموذج على التعميم.
تجدر الإشارة إلى أنه تم الاحتفاظ بصور خط خلية BJ في كل من مجموعة التدريب ومجموعة البيانات الاحتياطية 1.
قبل التجربة، قام فريق البحث بمعالجة الصور مسبقًا في HPA، والتي تضمنت الخطوات الخمس التالية ببساطة:
* الخطوة 1،تم تقليص حجم كل صورة 4 مرات، وتم تقليص الدقة النهائية إلى 0.32 ميكرومتر/بكسل من أجل تقليل كمية الحساب وإزالة الضوضاء عالية التردد؛
* الخطوة 2،تم الجمع بين التمويه الغاوسي (σ=5) وعتبة أوتسو لفصل المنطقة التقريبية لنواة الخلية عن الخلفية المعقدة؛
* الخطوة 3استخدم وظيفة remove_small_holes لإزالة الثقوب التي يقل حجمها عن 300 بكسل، ثم قم بتحويل الصورة إلى صورة ثنائية وإزالة مناطق الضوضاء التي يقل حجمها عن 100 بكسل؛
* الخطوة 4،تم حساب مركز كل نواة خلية، وتم اقتصاص مساحة 128 × 128 بكسل مع مركز الثقل كمنطقة اهتمام لخلية واحدة؛
* الخطوة 5،من خلال تطبيع الكثافة وتصفية الضوضاء، يتم تحقيق توزيع موحد للبيانات وتقليل التداخل بين القنوات.
هندسة النموذج: الجمع بين تسلسل البروتين وتمثيل الصور للتنبؤ بتوطين البروتين في الخلايا الفرعية
يتكون نموذج PUPS بشكل أساسي من جزأين:يتم استخدامه لتعلم تمثيل التسلسل من تسلسل الأحماض الأمينية للبروتين؛ يتم استخدام الآخر لتعلم تمثيل الصورة من خلال التلوين الأيقوني للخلية المستهدفة.يتم بعد ذلك الجمع بين تمثيل تسلسل البروتين وتمثيل الصورة للتنبؤ بالموقع الفرعي للبروتين في الخلايا المستهدفة. يتيح الأول تعميم النموذج على تنبؤات البروتين غير المعروفة، ويمكّن الأخير النموذج من التقاط التباين على مستوى الخلية الفردية وتحقيق تنبؤات توطين محددة لنوع الخلية. كما هو موضح في الشكل التالي:
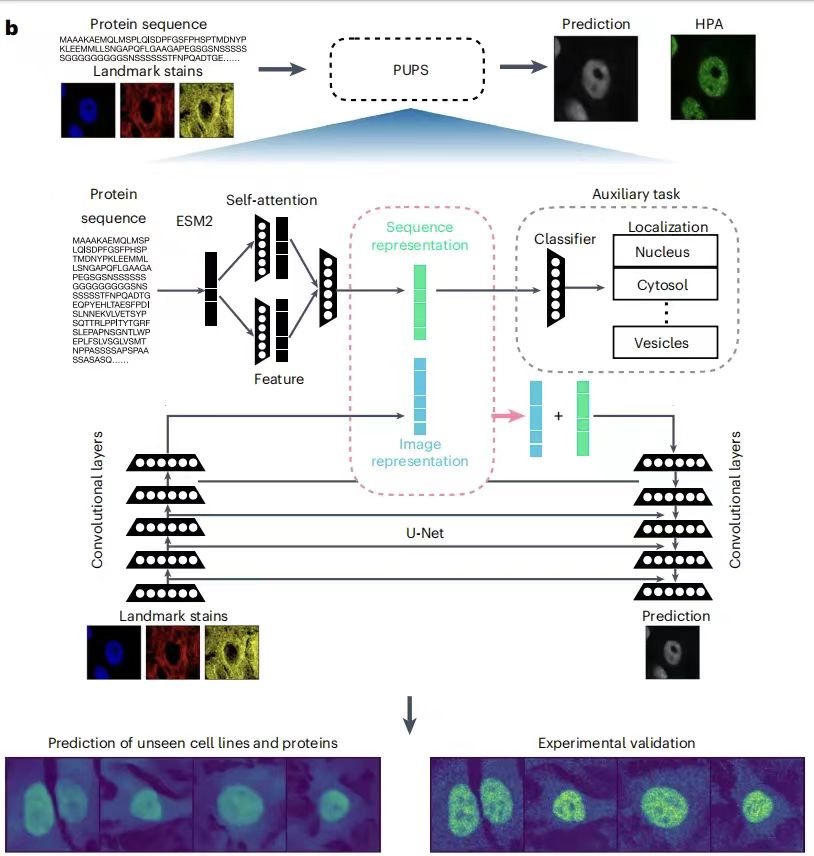
بعبارات بسيطة،يستخدم PUPS نموذج لغة البروتين ESM-2 (النمذجة التطورية على نطاق واسع) المدرب مسبقًا لاستخراج ميزات تسلسل البروتين، ويستخدم شبكة عصبية ملتوية لتعلم ميزات صورة التلوين الأيقونية للخلايا. وأخيرًا، يتم دمج الجزأين من المعلومات للتنبؤ بموقع البروتينات في الخلايا المستهدفة.تجدر الإشارة إلى أن جميع أجزاء النموذج يتم تدريبها في وقت واحد، مما يساعد على تقليل فقدان التصنيف للمهمة المسبقة والفرق بين صور البروتين المتوقعة وصور البروتين المقاسة تجريبياً في HPA. تم تحسين جميع المعلمات باستخدام محسن Adam بمعدل تعلم 1e-4.
نموذج لغة البروتين
يتعلم PUPS تمثيلات التسلسل باستخدام نموذج اللغة وطبقة الاهتمام الذاتي ومهمة التدريب المسبق المساعدة، ثم يقوم بتصنيف توطين البروتين بناءً على تمثيلات التسلسل التي تم تعلمها.
على وجه التحديد، حصل فريق البحث على تمثيل أولي لمتغير بروتيني محدد عن طريق إدخال تسلسل الأحماض الأمينية الطرفية N-2 الذي يحتوي على 2000 حمض أميني في نموذج ESM-2 المدرب مسبقًا، وبالتالي توليد متجه مكون من 1280 بُعدًا لكل بقايا حمض أميني، مع عدم وجود أي حشو للمتغيرات التي تحتوي على أقل من 2000 بقايا. يهدف قطع طول التسلسل هذا إلى تجنب التوقعات المتحيزة للبروتينات القليلة التي يصل طول تسلسلها إلى عشرات الآلاف من البقايا. كما هو موضح في الشكل التالي:
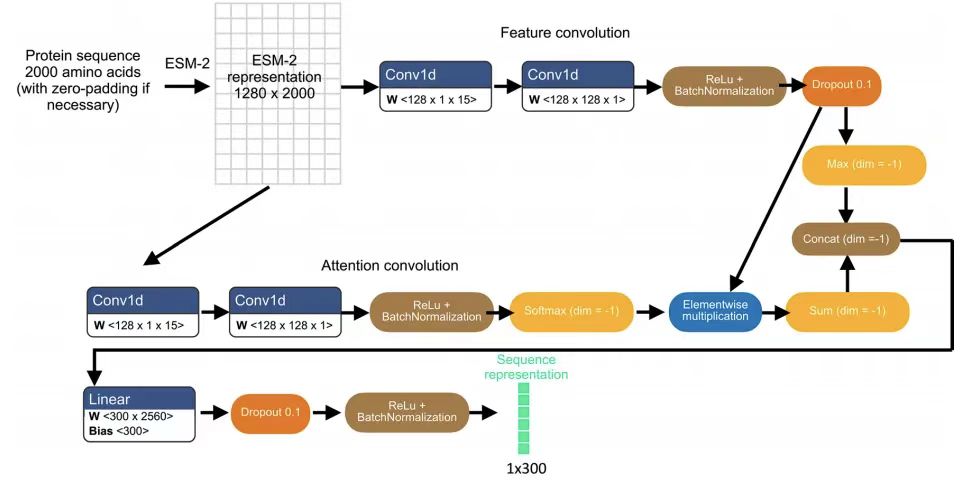
لتكييف خصائص ESM-2 للتنبؤ بموقع البروتين،وبعد ذلك، اعتمد الفريق طبقة انتباه خفيفة من التلافيف القابلة للفصل.عند تطبيقه على تمثيل ESM-2، يتم الحصول أخيرًا على تمثيل تسلسلي مكون من 300 بُعد. يتم استخدام تمثيل تسلسل البروتين هذا لكل من المهمة المسبقة المساعدة للتنبؤ بعلامات التوطين وللتنبؤ بصورة البروتين بالاشتراك مع تمثيل الصورة. تقوم المهمة المسبقة بإدخال تمثيل تسلسل البروتين في طبقة شبكة عصبية متصلة بالكامل لإدخال متجه مكون من 29 بُعدًا يمثل توزيع الاحتمالات بين 29 علامة توطين حجرة فرعية للخلية، ثم تقارن مخرجات المهمة المسبقة مع حجرات البروتين الموضحة بواسطة HPA باستخدام فقدان إنتروبيا متقاطع ثنائي مع تنشيط سيجما.
نموذج تقديم الصورة
يحتوي مدخل الصورة لكل خلية على ثلاث قنوات تلوين مميزة: نواة الخلية، والأنابيب الدقيقة، والشبكة الإندوبلازمية.أبعادها هي 3 × 128 × 128 ومركزها هو مركز النواة.
يتم ترميز الصورة من خلال 5 طبقات ملتوية قابلة للفصل.الأبعاد النهائية 16 × 16 × 512. يتم متابعة كل طبقة ملتوية من خلال تنشيط leakyRelu، وتطبيع الدفعات، وطبقات التجميع 2D max. يتم ربط تمثيل تسلسل البروتين بجميع الأبعاد المكانية لتمثيل صورة الخلية ثم إدخاله في فك تشفير صورة U-Net الذي يتعلم أوزانًا مختلفة لكل قناة إدخال. بالإضافة إلى ذلك، تسمح آلية ترجيح الأبعاد المكانية في النموذج بدمج كل بُعد مكاني لتمثيل الصورة مع تمثيل التسلسل بأوزان مختلفة.
يتكون فك التشفير من 5 طبقات ملتوية قابلة للفصل.يقوم بإنشاء إخراج صورة بحجم 1 × 128 × 128، وهو عبارة عن تنبؤ بصورة البروتين للخلية المقابلة. ثم يتم إضافة اتصالات تخطي مماثلة لتلك الموجودة في تقسيم الصورة U-Net بين طبقة التشفير التي تولد تمثيلات الصور للتلوين المعلمي وطبقة فك التشفير التي تولد تنبؤات صور البروتين على نفس العمق. استخدمت الدراسة دالة خسارة الخطأ التربيعي المتوسط لتدريب النموذج لتقليل الفرق بين صور البروتين المتوقعة وصور البروتين المقاسة تجريبياً.
النتائج التجريبية: تحقيق توطين دقيق للبروتينات على مستوى الخلية الواحدة
ومن أجل التحقق من جدوى وفعالية النموذج، اقترح فريق البحث إجراء عدد من التجارب للتحقق. أظهر PUPS أداءً جيدًا في مهام متعددة، مما يسلط الضوء على مزاياه في اندماج النماذج المتعددة.
التنبؤ بالتباين في توطين البروتين بين خطوط الخلايا
لتقييم أداء PUPS في تحديد كمية التباين في توطين البروتين بين خطوط الخلايا،قام فريق البحث بحساب نسبة النواة البروتينية لقياس تباين التوطين ووجد أن القيم المتوقعة كانت مرتبطة بشكل كبير بالبيانات الحقيقية.معامل ارتباط بيرسون للحالة الأولى هو 0.794، ومعامل ارتباط بيرسون للحالة الثانية هو 0.878. كما هو موضح في الشكل التالي:
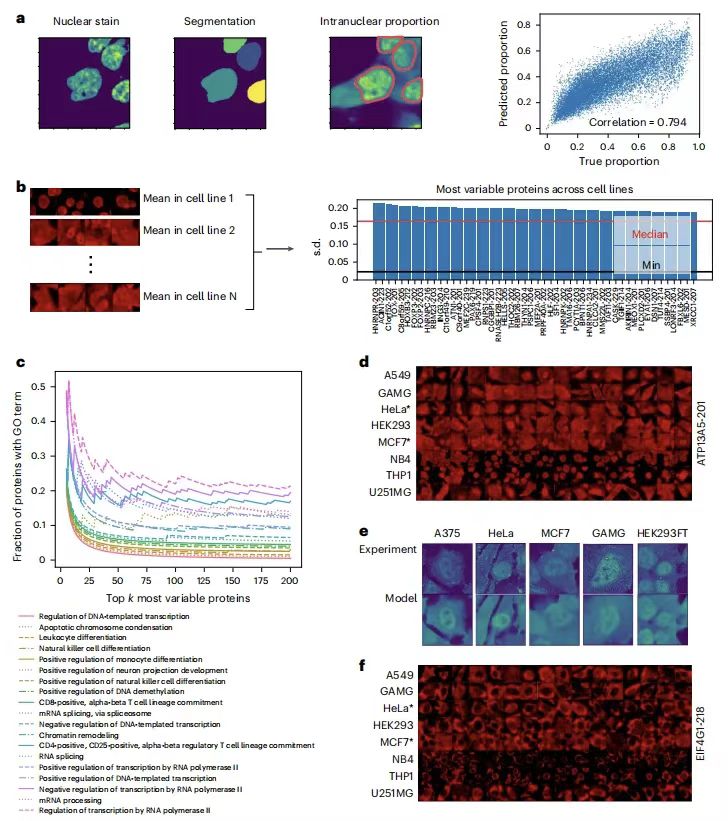
وأظهر التحليل الإضافي اللاحق أن البروتينات ذات أكبر تغيرات التوطين بين خطوط الخلايا كانت مرتبطة بالعمليات البيولوجية مثل النسخ، وتمايز الخلايا، وتنظيم الكروماتين. وقد أكد التحقق التجريبي من صحة ATP13A5 دقة توقعات النموذج. أيضًا،يلتقط النموذج الاختلافات في مورفولوجيا الخلايا من خلال تلطيخ التوقيع ويمكنه استنتاج خصوصية خط الخلية من خلال توطين البروتين دون تسميات خط الخلية، مما يوفر طريقة جديدة لدراسة التنظيم الخاص بالخلية لوظيفة البروتين.
التنبؤ بالاختلافات في توطين البروتين بين الخلايا الفردية
لتقييم قدرة PUPS على التنبؤ بتباين توطين البروتين بين الخلايا الفردية في نفس خط الخلية، قام فريق البحث بحساب تباين النسبة النووية للبروتينات في جميع الخلايا الفردية في كل خط خلية.وأظهرت النتائج أن تصنيف التنبؤ بتباين الخلية الفردية لكل زوج من البروتينات وخطوط الخلايا كان متسقًا للغاية مع البيانات الحقيقية.على سبيل المثال، تجاوز معدل التداخل لأول 500 زوج من المتغيرات العالية في Holdout 2 معدل 60%، وكان توزيع النسبة النووية الداخلية المتوقع متوافقًا مع النتائج الفعلية، مما أدى إلى القضاء على تأثير أخطاء التنبؤ.
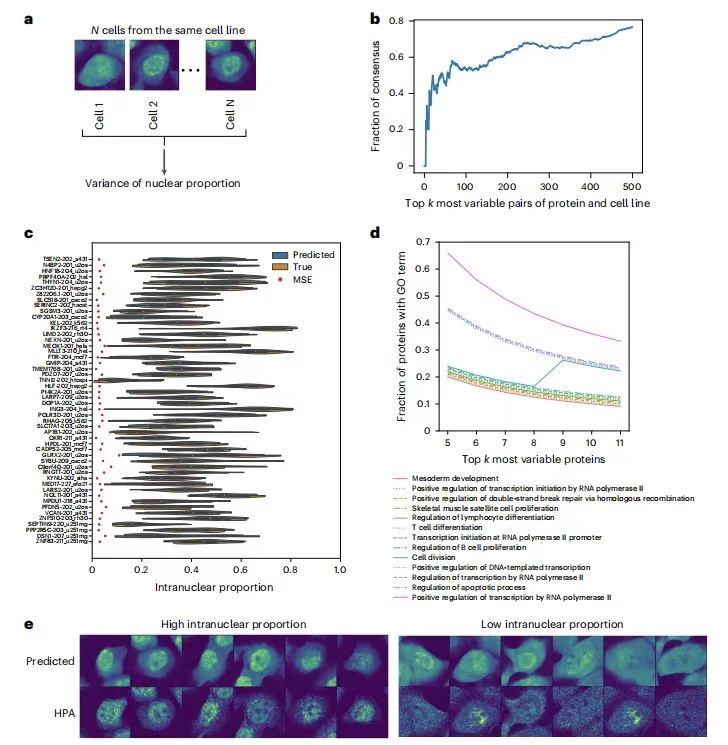
بالإضافة إلى ذلك، أظهر تحليل علم الجينات (GO) أن البروتينات شديدة التغير كانت مرتبطة بعمليات مثل انقسام الخلايا، والنسخ، وإصلاح الكسر المزدوج، والاستماتة. أيضًا،يلتقط النموذج السمات المورفولوجية من خلال صور صبغ الخلايا، مما يشير إلى أن التباين في الخلية الفردية ليس عشوائيًا فحسب، بل يرتبط أيضًا بالسمات المورفولوجية للخلايا.يقدم منظورًا جديدًا لشرح آلية التباين في الخلية الواحدة.
التحقق من صحة PUPS في التجارب الجديدة خارج بيانات التدريب
للتحقق من قدرة PUPS على التنبؤ بتوطين البروتين في بيئات تجريبية جديدة، اختار فريق البحث 9 بروتينات للتحقق منها في 5 خطوط خلوية. كما هو موضح في الشكل التالي:
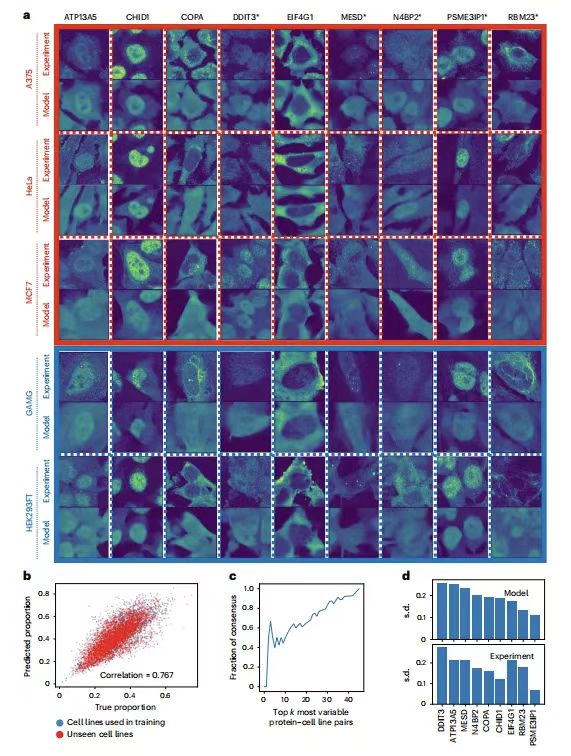
ATP13A5، CHID1، COPA، MESD وRBM23 هي البروتينات التي تحتوي على أكبر قدر من التنوع بين خطوط الخلايا، وكلها لها مصطلحات GO مختلفة؛ DDIT3 و N4BP2 هما البروتينان اللذان يتمتعان بأكبر قدر من التنوع في الخلايا الفردية ضمن سلالة الخلية؛ يعد EIF4G1 وPSME3IP1 من البروتينات التي تحتوي على أقل قدر من التنوع بين خطوط الخلايا، ومن المتوقع أن يقع الأول بشكل رئيسي خارج النواة، ومن المتوقع أن يقع الأخير بشكل رئيسي داخل النواة. من بين خطوط الخلايا الخمسة، باستثناء A375، تم تضمين HeLa الأخرى، MCF7، GAMG وHEK293FT في HPA.
وتظهر النتائج أنإن صور البروتين التي تنبأت بها PUPS متشابهة بصريًا مع تلك التي تم قياسها تجريبيًا.إن نسبة البروتين النووي لكل خلية مفردة، المحسوبة باستخدام صورة البروتين المتوقعة، ترتبط ارتباطًا وثيقًا بالنسبة المحسوبة من الصورة المقاسة تجريبيًا، مع معامل ارتباط بيرسون 0.767. وهذا يدل على أنيمكن استخدام PUPS للتنبؤ كميًا بموقع البروتينات التي لم يتم قياسها تجريبيًا أو استخدامها في أطالس التدريب مسبقًا.
يتعلم PUPS تمثيلات البروتين والخلايا ذات المغزى
تثبت التجارب أن قدرة PUPS على التنبؤ بموقع البروتين في البروتينات وخطوط الخلايا غير المعروفة تأتي من تعلم التمثيلات ذات المغزى لتسلسلات البروتين وصور المعالم الخلوية.
قام فريق البحث برسم خريطة لتمثيلات تسلسل البروتين لـ 40,622 شكلًا بروتينيًا يتوافق مع 12,614 جينًا، وتميل البروتينات ذات التوطينات المماثلة إلى أن يكون لها تمثيلات تسلسل مماثلة. ولإثبات أن النموذج يمكنه تحديد أنماط تسلسل البروتين ذات المغزى والتنبؤ بالتوطين، استخدم فريق البحث طريقة Shapley الموضعية لحساب أهمية كل بقايا الأحماض الأمينية في بروتين معين للتنبؤ بعلامات كل حجرة خلوية. على سبيل المثال، فقد أوضح بنجاح التباين المتوقع في توطين N4BP2 النووي، وهو ما يتفق مع التقارير التي تفيد بأن مجال CUE قد يغير توطين الخلايا الفرعية من خلال ربط اليوبيكويتين.
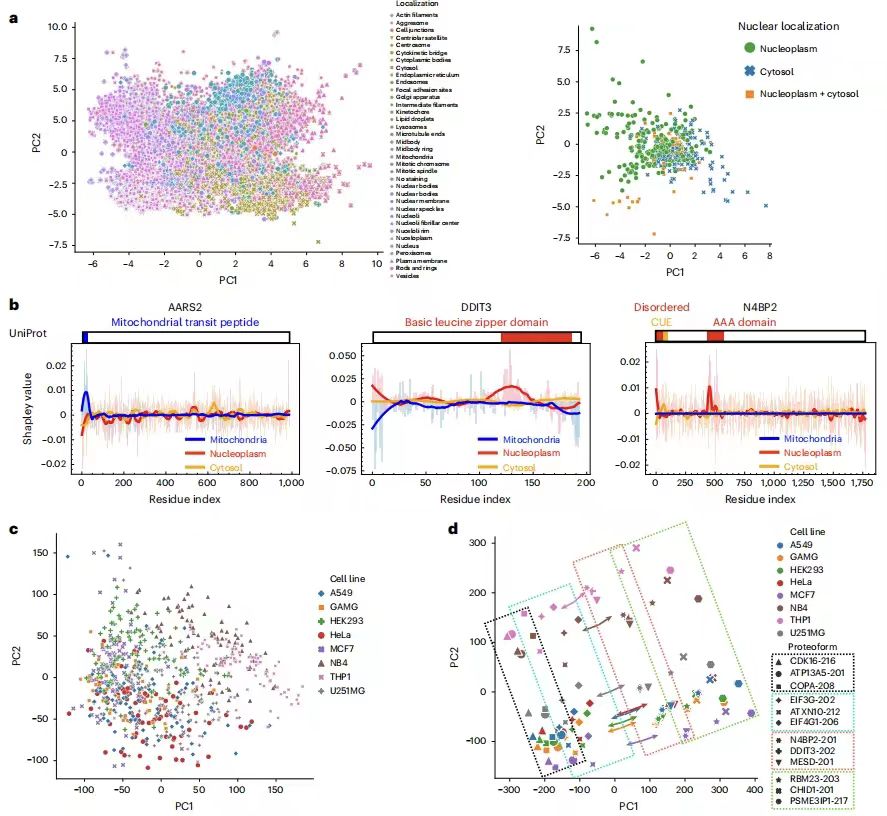
بالإضافة إلى تحديد أنماط تسلسل البروتين ذات المعنى،وأظهر فريق البحث أيضًا أن PUPS يتعلم تمثيلات ذات معنى للخلايا الفردية من خلال تلطيخ التوقيع الخلوي.يقوم بتصور تمثيلات صور الخلايا الفردية التي تم تعلمها من تلطيخ المعالم ويجد أن الخلايا الفردية من نفس خط الخلية لها تمثيلات صور مماثلة حتى لو لم يتم إدخال تسمية خط الخلية في النموذج. يحافظ التمثيل المشترك للبروتينات وصور المعالم الخلوية على الفصل بين خطوط الخلايا والبروتينات، في حين يكون ترتيب البروتينات المختلفة داخل كل خط خلوي متشابهًا عبر خطوط الخلايا. بالنظر إلى مركز ثقل كل خط خلوي في مساحة التمثيل المشترك، فإن المتجهات من مركز ثقل كل خط خلوي إلى بروتين محدد تكون متوازية في الغالب عبر جميع خطوط الخلايا، أي أنه بالنظر إلى تمثيل التسلسل، فإن التنبؤ بصورة لبروتين محدد يتطلب التحرك في نفس الاتجاه في مساحة التمثيل بغض النظر عن خط الخلية.وهذا يفسر قدرة PUPS على التعميم على البروتينات وخطوط الخلايا غير المعروفة من خلال تعلم التمثيلات ذات المغزى لصور البروتين والخلايا.
أيضًا،يمكن لـ PUPS أيضًا التنبؤ بتأثيرات الطفرات المسببة للأمراض على توطين البروتين.على سبيل المثال، أظهرت دراسات الطفرات على البروتينات الميتوكوندريا المشفرة نوويًا SDHD و ETHE1 أن طفرات SDHD تؤدي إلى زيادة نسبة توطينها النووي، وهو ما يتوافق مع آلية عدم استقرار الجينوم النووي في المرض؛ تظهر طفرات ETHE1 زيادة في نسبة التوطين السيتوبلازمي، والتي ترتبط بتشوهات النقل النووي السيتوبلازمي المعروفة. تشير هذه النتائج إلى أن PUPS يمكن أن توفر أدلة جديدة لدراسة آليات المرض من خلال تحليل آثار اختلاف التسلسل على التوطين.
حل جديد للتنبؤ بتوطين البروتين تحت الخلوي
كما ذكر أعلاه، فإن التنبؤ بتوطين البروتين تحت الخلوي له أهمية كبيرة في كل من البحث الحيوي والبحث البيولوجي. يوفر برنامج PUPS طريقة لدمج المعلومات المتعددة الوسائط، مما يضيف لمسة قوية إلى البحث في هذا المجال. وفي الوقت نفسه، وبعد عقود من التطوير، أسفرت الأبحاث في هذا المجال عن مجموعة واسعة من النتائج.
نشر فريق من جامعة كلية دبلن في أيرلندا دراسة في مجلة Computational and Structural Biotechology.يقدم هذا الكتاب مجموعة متنوعة من الأساليب الحسابية للتنبؤ بتوطين البروتين تحت الخلوي، بما في ذلك الأساليب القائمة على التسلسل، والأساليب القائمة على الشرح التوضيحي، والأساليب الهجينة، والأساليب القائمة على التنبؤ التلوي. كما تقوم المقالة أيضًا بتصنيف وتقديم أدوات التنبؤ بالتوطين الفرعي للخلايا حسب حقيقيات النوى، وبدائيات النوى، والفيروسات، وفئات متعددة.تتضمن أدوات التنبؤ حقيقية النواة mLASSO-Hum وDeepPSL وما إلى ذلك، وتتضمن أدوات التنبؤ بدائية النواة PRED-LIPO وما إلى ذلك. من خلال تصميم خريطة تصنيف للتعلم الآلي والتعلم العميق تغطي 7 مجالات رئيسية و28 فئة فرعية، توفر هذه الدراسة تصنيفًا لأدوات التنبؤ أحادية الفئة ومتعددة الفئات، مما يسهل على المستخدمين العثور على الأساليب وأدوات التنبؤ. نُشرت الورقة البحثية تحت عنوان "أدوات التنبؤ بتوطين البروتين الفرعي الخلوي".
* عنوان الورقة:
https://www.sciencedirect.com/science/article/pii/S2001037024001156
في 12 أبريل، تعاونت مجموعة أبحاث يانغ لي في معهد العلوم الطبية الحيوية بجامعة فودان ومجموعة أبحاث دونغ نانكينغ في مختبر الذكاء الاصطناعي في شنغهاي، لنشر ورقة بحثية عبر الإنترنت بعنوان "نموذج توليدي عميق لتوطين البروتين الفرعي الخلوي" في مجلة Briefings in Bioinformatics.كما طورت الدراسة نموذج التعلم العميق التوليدي deepGPS مع قدرات معالجة متعددة الوسائط تعتمد على نموذج لغة البروتين ESM2 وإطار عمل U-Net.
وفقًا للتقارير، يمكن لـ deepGPS استقبال تسلسلات البروتين وصور نواة الخلية كمدخلات وإنشاء تسميات نصية وصور توزيع لموقع البروتين. إنه نموذج جديد متعدد الوسائط "من النص إلى الصورة" يدعم التنبؤ بتوطين البروتين تحت الخلوي.
* عنوان الورقة:
https://doi.org/10.1093/bib/bbaf152
مع تسارع تكامل الذكاء الاصطناعي والبحث البيولوجي، تظهر باستمرار تجارب مبتكرة ذات صلة، تكسر تدريجيا عيوب الطرق التقليدية، وتحقق "أفضل ما في العالمين" أو حتى الأداء "المثالي"، وبالتالي تعزيز التطور السريع لعلم المعلومات الحيوية.








