Command Palette
Search for a command to run...
قام فريق Zhang Yang في الجامعة الوطنية في سنغافورة بتطوير خوارزمية للتنبؤ ببنية الحمض النووي الريبي من الجيل الثاني والتي تفوقت على SOTA في اختبارات معيارية متعددة

لقد كان فهم بنية ووظيفة جزيئات الحمض النووي الريبي دائمًا اتجاهًا بحثيًا أساسيًا في علم الأحياء الجزيئي وصناعة الأدوية. يمكن للحمض النووي الريبوزي (RNA)، وخاصة الحمض النووي الريبوزي غير المشفر (ncRNA)، أن يتحول إلى هياكل محددة ويلعب أدوارًا مهمة في العديد من العمليات الخلوية مثل تنظيم الجينات (مثل النسخ والترجمة)، والتحفيز، ونقل الإشارة البيولوجية، والاستجابة للإجهاد.
مع التطور السريع لتكنولوجيا التسلسل عالية الإنتاجية، نمت بيانات تسلسل الحمض النووي الريبي بشكل كبير، لكن الفجوة بين التسلسلات المعروفة وهياكل الحمض النووي الريبي التي تم حلها تجريبياً تتسع. ومن ثم، أصبح من الضروري بشكل متزايد حل البنية الذرية للحمض النووي الريبي استناداً فقط إلى تسلسلها الخام. قام الباحثون بتطوير مجموعة متنوعة من الأساليب لدراسة بنية الحمض النووي الريبي، مثل تقنيات علم الأحياء البنيوي مثل علم البلورات بالأشعة السينية، والتصوير بالرنين المغناطيسي النووي، والمجهر الإلكتروني بالتبريد (cryo-EM). على الرغم من أن هذه التقنيات التجريبية يمكن أن توفر دقة أعلى، فإن التوضيح التجريبي للبنية ثلاثية الأبعاد للحمض النووي الريبي غالبًا ما يكون مكلفًا، وفي بعض الحالات، يصعب تحقيقه. لذلك،هناك طلب متزايد على الأساليب الحسابية للتنبؤ ببنية الحمض النووي الريبي ثلاثية الأبعاد عالية الجودة مباشرة من التسلسل.
يشير مصطلح "التنبؤ ببنية الحمض النووي الريبي من البداية" إلى طريقة تتنبأ بشكل مباشر بالبنية ثلاثية الأبعاد للحمض النووي الريبي من تسلسلها دون الاعتماد على أي بيانات تجريبية أو معرفة مسبقة. يعتمد جوهر هذه الطريقة على استخدام محاكاة الكمبيوتر وتقنيات الكيمياء الحسابية للتنبؤ بالتكوين الثلاثي الأبعاد لجزيئات الحمض النووي الريبي من خلال النماذج الرياضية والخوارزميات.
في الآونة الأخيرة، أدت أحدث نتائج الأبحاث التي أجراها فريق البروفيسور تشانغ يانغ في الجامعة الوطنية في سنغافورة إلى تعزيز "التنبؤ ببنية الحمض النووي الريبي من النوع Ab initio" إلى مستوى أعلى.اقترح الباحثون إطار عمل عالي الدقة للتنبؤ ببنية الحمض النووي الريبي (RNA) يعتمد على التعلم العميق، DRfold2.إنه يدمج نموذج لغة RNA المركب المدرب مسبقًا (RCLM) ووحدة بنية إزالة الضوضاء للتنبؤ ببنية RNA من البداية إلى النهاية. يحقق DRfold2 أداءً جيدًا في كل من الطوبولوجيا العالمية والتنبؤ بالهيكل الثانوي مقارنةً بالطرق الحديثة الأخرى على معايير متعددة.
يُظهر التحليل التفصيلي أن هذا التحسن يأتي بشكل أساسي من قدرة RCLM على التقاط الأنماط التطورية المشتركة وعملية إزالة الضوضاء الفعالة.يؤدي هذا إلى تحسين دقة التنبؤ بالاتصال غير الخاضع للإشراف لـ DRfold2 بما يزيد عن 100% مقارنة بالطرق الحالية.
وقد تم نشر النتائج ذات الصلة على منصة الطباعة المسبقة bioRxiv تحت عنوان "التنبؤ ببنية RNA من البداية باستخدام نموذج اللغة المركبة والتعلم الشامل الخالي من الضوضاء".
أبرز الأبحاث:
* يدمج DRfold2 نموذج لغة RNA المركب المدرب مسبقًا (RCLM) ووحدة بنية إزالة الضوضاء للتنبؤ ببنية RNA من البداية إلى النهاية
* من خلال مزيج فريد من نمذجة اللغة المركبة، والتعلم الشامل القائم على إزالة الضوضاء، والتحسين اللاحق الموجه بالتعلم العميق، يفتح DRfold2 اتجاهًا جديدًا لـ "التنبؤ ببنية الحمض النووي الريبي من البداية"
* يُعد DRfold2 مكملًا للغاية لـ AlphaFold3 ويحقق تحسينات كبيرة في الدقة إحصائيًا بعد التكامل في إطار التحسين

عنوان الورقة:
https://www.biorxiv.org/content/10.1101/2025.03.05.641632v1
تنزيل مجموعة بيانات اختبار بنية RNA الخاصة بـ DRfold2:
مجموعة البيانات: إنشاء مجموعة بيانات اختبار مستقلة
من أجل تقييم أداء DRfold2 بشكل موضوعي،قام الباحثون ببناء مجموعة بيانات اختبار مستقلة تحتوي على 28 بنية من الحمض النووي الريبي.أطوال تسلسلها كلها أقل من 400 nts وتأتي من الفئات الثلاث التالية:
* أحدث تسلسلات أهداف RNA-Puzzles
* تسلسلات هدف الحمض النووي الريبي في مسابقة CASP15
* أحدث هياكل الحمض النووي الريبي المنشورة في قاعدة بيانات بنك بيانات البروتين (PDB) اعتبارًا من 1 أغسطس 2024
ومن الجدير بالذكر أن الباحثين استبعدوا هياكل الحمض النووي الريبي الاصطناعي الكبيرة من مجموعة بيانات CASP15 لأنها تنحرف عن هياكل الحمض النووي الريبي الموجودة في الطبيعة، والتي تشكل التركيز الأساسي للتحليل الوظيفي وتصميم الأدوية.
ولضمان تقييم دقيق للنموذج، تحتوي مجموعة التدريب فقط على هياكل الحمض النووي الريبي المنشورة قبل عام 2024، وتستبعد الحمض النووي الريبي الذي يتشابه تسلسله مع مجموعة بيانات الاختبار بما يزيد عن 80%.
تنزيل مجموعة بيانات اختبار بنية RNA الخاصة بـ DRfold2:
هندسة النموذج: خط أنابيب جديد للتنبؤ ببنية الحمض النووي الريبي ثلاثي الأبعاد DRfold2
DRfold2 هو خط أنابيب جديد للتنبؤ ببنية RNA ثلاثية الأبعاد يتكون من أربع وحدات أساسية: (1) نموذج لغة RNA المركبة (RCLM)، (2) كتلة محول RNA، (3) وحدة بنية إزالة الضوضاء، و(4) اختيار النموذج النهائي وتحسينه من خلال بروتوكول CSOR، كما هو موضح في الشكل أ أدناه:
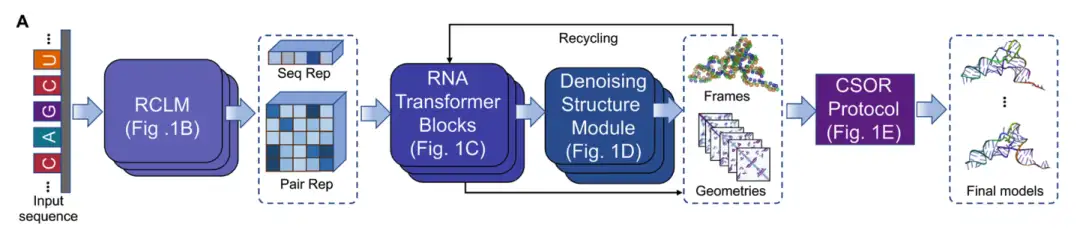
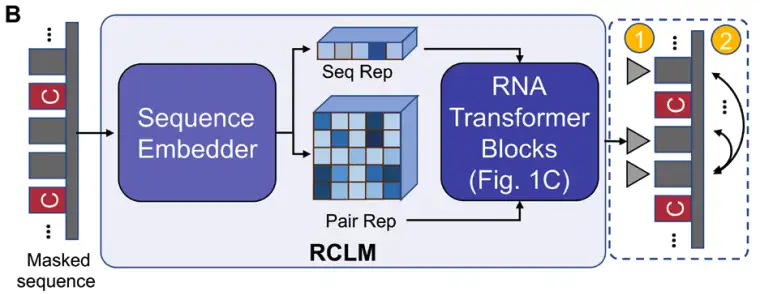
بدءًا من تسلسل RNA المدخل،يقوم DRfold2 أولاً بترميز تسلسل الاستعلام باستخدام نموذج لغة مركب RNA مدرب مسبقًا (RCLM).إنشاء تمثيل التسلسل (Seq Rep) وتمثيل الزوج (Pair Rep)؛ يتم تدريب RCLM على بيانات تسلسل غير خاضعة للإشراف على نطاق واسع من خلال طريقة تعظيم الاحتمالية المركبة لتحقيق التعرف على نمط التسلسل بشكل أكثر كفاءة، كما هو موضح في الشكل B أدناه:
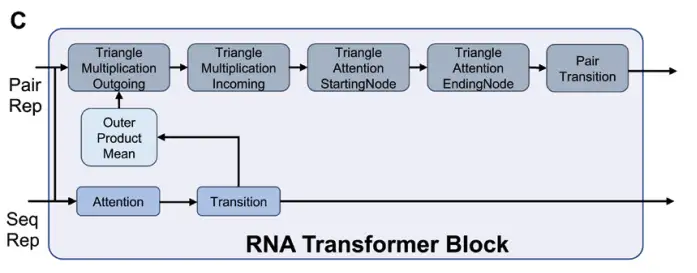
يتم بعد ذلك إدخال هذه التسلسلات والتمثيلات المزدوجة في وحدة محول الحمض النووي الريبي للمعالجة من أجل توليد تمثيلات الميزات الرئيسية المطلوبة لطي بنية الحمض النووي الريبي، كما هو موضح في الشكل C أدناه:
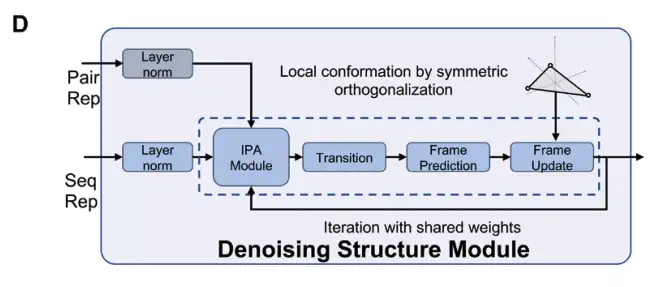
بعد ذلك، يستخدم DRfold2 وحدة بنية الحمض النووي الريبي لإزالة الضوضاء (DRSM) لتوليد تكوينات الحمض النووي الريبي بطريقة شاملة، كما هو موضح في الشكل D أدناه:
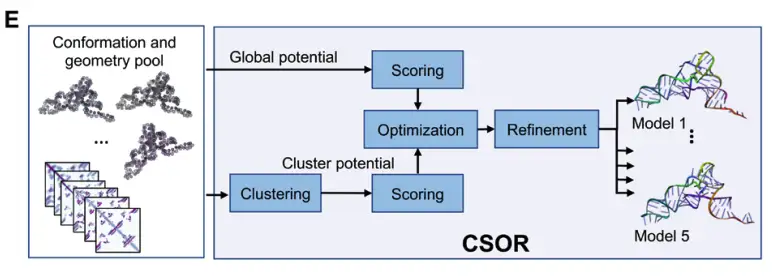
يتم فحص نموذج بنية الحمض النووي الريبي النهائي وتحسينه من خلال بروتوكول CSOR بعد المعالجة لتحديد وتنقية أفضل نموذج من مجموعة التكوينات التي تم إنشاؤها عند نقاط تفتيش متعددة، كما هو موضح في الشكل E أدناه:
على الرغم من أن DRfold2 يحمل اسمًا مشابهًا لطريقة DRfold السابقة التي ابتكرها الفريق، إلا أنه يقدم تقدمًا كبيرًا استنادًا إلى إطار عمل مختلف تمامًا.الأمر الأكثر أهمية هو دمج نموذج اللغة المركبة، والذي يعزز بشكل كبير قدرة تسلسل الحمض النووي الريبي وتمثيل الأزواج.بالإضافة إلى ذلك، يدمج خط أنابيب التنبؤ وحدة بنية الحمض النووي الريبي لإزالة الضوضاء (DRSM)، والتي تستخدم استراتيجية اضطراب محكومة لتعلم التحولات البنيوية بشكل قوي من خلال تصحيح تكوينات الحمض النووي الريبي الضوضائية بكفاءة.
قام الباحثون بإتاحة خادم DRfold2 عبر الإنترنت والرمز المحلي للعامة على:
https://zhanglab.comp.nus.edu.sg/DRfold2
نتائج البحث: DRfold2 يتفوق على الطرق الحديثة الأخرى في معايير متعددة
قام الباحثون أولاً بمقارنة DRfold2 مع خمسة طرق متطورة للتنبؤ ببنية الحمض النووي الريبي، بما في ذلك RNAComposer (تجميع الأجزاء وتحسينها)، وtrRosettaRNA (طريقة التعلم العميق)، وRhoFold (طريقة التعلم العميق من البداية إلى النهاية)، وRoseTTAFoldNA (طريقة التعلم العميق من البداية إلى النهاية) وDeepFoldRNA (طريقة التعلم العميق).
كما هو موضح في الشكل أدناه، قارن الباحثون نتائج تقييم TM-score وRMSD لـ DRfold2 وطريقة المعيار عند عتبات تشابه التسلسل المختلفة (50%-80%). ومن بينها، TM-score هي وظيفة تسجيل مستقلة عن الطول تستخدم لتقييم الجودة الشاملة لبنية RNA المتوقعة. نطاق القيمة هو 0-1. كلما زادت القيمة، كلما زاد التشابه بين البنية المتوقعة والبنية الحقيقية.
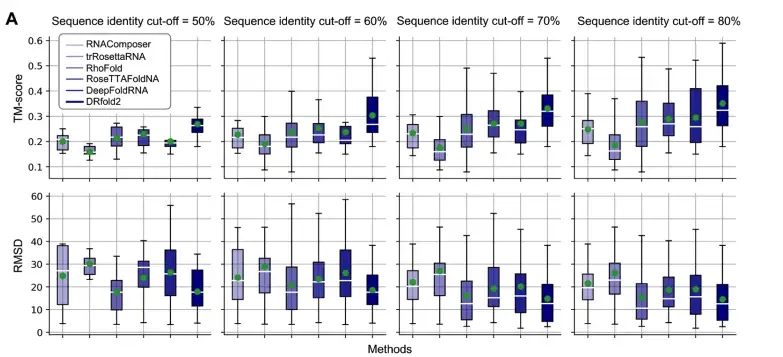
تظهر النتائج أن DRfold2 يحصل دائمًا على أعلى متوسط درجة TM في ظل جميع عتبات تشابه التسلسل.على سبيل المثال:
* تحت عتبة التشابه 80%، فإن متوسط درجة TM لـ DRfold2 هو 0.351، وهو أعلى بمقدار 18.6% من DeepFoldRNA الذي يحتل المرتبة الثانية (درجة TM=0.296).
* تحت عتبة التشابه 50% (مجموعة الاختبار الأكثر صرامة)، لا يزال بإمكان DRfold2 الحصول على متوسط درجة TM يبلغ 0.269، وهو أعلى بمقدار 17.5% من RoseTTAFoldNA الذي يحتل المرتبة الثانية (درجة TM=0.229).
* بالإضافة إلى ذلك، فإن RMSD (الانحراف الجذري المتوسط التربيعي) لـ DRfold2 عند جميع عتبات تشابه التسلسل يكون دائمًا أقل من كل طرق التحكم، مما يشير إلى أن بنيته المتوقعة أقرب إلى بنية RNA الحقيقية.
كما استخدم الباحثون أيضًا الريبوزيم الشبيه بـ CPEB3 HDV الموجود في الشمبانزي (معرف قاعدة بيانات البروتينات: 7QR3) كمثال. يبلغ طول الحمض النووي الريبوزي RNA 69 نوكليوتيدًا وتم تحليل تأثيرات التنبؤ بالطرق المختلفة على البنية الثلاثية للحمض النووي الريبوزي RNA. وكانت النتائج على النحو التالي:
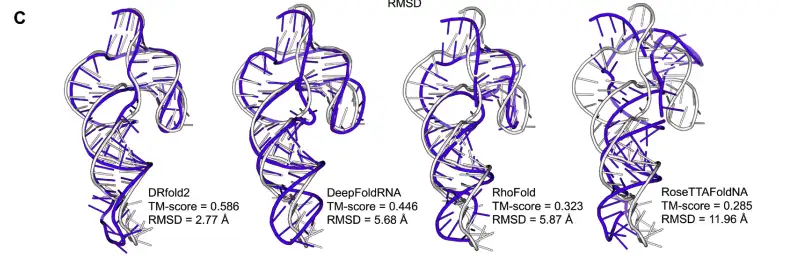
* التقط DRfold2 بدقة التركيب الطوبولوجي الكلي للريبوزيم، مع درجة TM تبلغ 0.586 وانحراف معياري متوسط التربيع يبلغ 2.77 Å فقط.
* يؤدي DeepFoldRNA أداءً جيدًا من حيث الترتيب الحلزوني الشامل، ولكن اتجاه حلقة دبوس الشعر ينحرف بشكل كبير، مما يؤدي إلى RMSD يصل إلى 5.68 Å، وهو ضعف انحراف DRfold2.
* RhoFold وRoseTTAFoldNA لديهما أخطاء أكبر في التنبؤ المكاني في مناطق التقاطع، مما يتسبب في انخفاض درجة TM إلى 0.323 و0.285.
* أعلى تشابه في التسلسل بين RNA المستهدف ومجموعة البيانات التدريبية هو 60.9% فقط، مما يشير إلى أن DRfold2 لا يزال بإمكانه توفير تنبؤات هيكلية موثوقة لتسلسلات RNA الجديدة في غياب القوالب المتجانسة.
وتظهر هذه النتائج أن:إن التمثيل الاحتمالي الشامل الذي توفره نماذج اللغة ذات الترتيب الأعلى مثل RCLM يعزز بشكل كبير القدرة على تعلم الأنماط المتطورة والقيود المكانية.وبالتالي، تم تحقيق نمذجة أكثر دقة لبنية الحمض النووي الريبي ثلاثي الأبعاد من خلال الشبكة الشاملة لـ DRfold2.
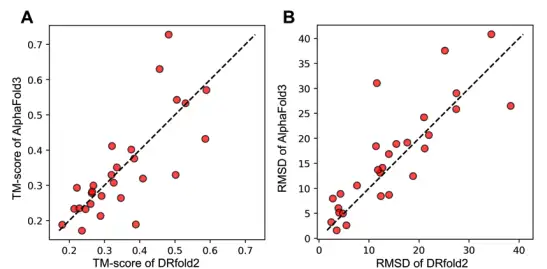
وعلى هذا الأساس، من أجل مقارنة أداء DRfold2 وAlphaFold3 في التنبؤ ببنية RNA ثلاثية الأبعاد، أرسل الباحثون أيضًا تسلسلات RNA في مجموعة الاختبار إلى خادم AlphaFold وحصلوا على البنية المتوقعة لـAlphaFold3 باستخدام تكوين البذرة الافتراضي. تحول،متوسط درجة TM (0.351) والانحراف المعياري الجذري المتوسط (14.6 Å) لـ DRfold2 أعلى قليلاً من تلك الخاصة بـ AlphaFold3 (0.345 و16.0 Å).
الأمر الأكثر أهمية هو أنه على الرغم من أن DRfold2 وAlphaFold3 يظهران أداءً عامًا مشابهًا، فإن النتائج في الشكل أدناه تسلط الضوء على التكامل القوي بين الاثنين، خاصةً عندما ينحرف التنبؤ بشكل كبير عن الخط القطري.من خلال دمج تنبؤات AlphaFold3 كمصطلح وظيفة محتمل إضافي في إطار تحسين DRfold2، حقق الباحثون تحسينات كبيرة إحصائيًا في كل من درجة TM وRMSD.
ركز فريق البروفيسور تشانغ يانغ على أبحاث الذكاء الاصطناعي وعلم الأحياء الحسابي لسنوات عديدة
إن نموذج DRfold2 المقترح في هذه الدراسة هو في الواقع نسخة مطورة من نموذج DRfold الذي اقترحه فريق البروفيسور Zhang Yang سابقًا.
في سبتمبر 2023، نشر فريق البروفيسور تشانغ يانغ ورقة بحثية بعنوان "دمج التعلم الشامل مع الإمكانات الهندسية العميقة للتنبؤ ببنية الحمض النووي الريبي من البداية" في مجلة Nature Communications.
تتحدث هذه الدراسة عن تقنية جديدة، DRfold، للتنبؤ الدقيق بالبنية ثلاثية الأبعاد للحمض النووي الريبي.يكمن الابتكار الأساسي في تقديم وظيفتين متكاملتين للطاقة الكامنة: إمكانات FAPE والإمكانات الهندسية.يتم تدريبهم من خلال شبكتي محول مستقلتين ويشكلان معًا إمكانات التعلم العميق للتنبؤ ببنية الحمض النووي الريبي. تظهر النتائج الحسابية أنه بالمقارنة مع طرق التنبؤ الحاسوبية السابقة لبنية الحمض النووي الريبي، فإن DRfold يتفوق على هذه الطرق في العديد من مؤشرات الأداء.

عنوان الورقة:
https://www.nature.com/articles/s41467-023-41303-9
من DRfold إلى DRfold2، واصل فريق البروفيسور تشانغ يانغ التركيز على أبحاث الذكاء الاصطناعي وعلم الأحياء الحسابي لسنوات عديدة. يعد مختبره أحد أوائل المختبرات التي تقوم بإجراء أبحاث التنبؤ ببنية البروتين والحمض النووي الريبي استنادًا إلى التعلم الآلي العميق. وقد فازت بجوائز مثل جائزة سلون الأمريكية، وجائزة المؤسسة الوطنية للعلوم في الولايات المتحدة، وجائزة الأبحاث العلمية الأساسية من جامعة ميشيغان. منذ عام 2015، تم اختيارها في قائمة Thomson Reuters/Clarivate Analytics لأكثر العلماء استشهاداً على مستوى العالم سبع مرات. خوارزمية I-TASSER التي طورها مختبره (https://zhanggroup.org/I-TASSER/), منذ عام 2006، تم تصنيفها باعتبارها الطريقة الأكثر دقة للتنبؤ ببنية البروتين الآلية في تجارب CASP العالمية لمدة تسع مرات متتالية.
في 2 يناير 2024، نشر فريق البروفيسور تشانغ يانغ ورقة بحثية بعنوان "تحسين التنبؤ بمونومر بروتين التعلم العميق والبنية المعقدة باستخدام DeepMSA2 مع بيانات ميتاجينوميات ضخمة" في مجلة Nature Methods.
طورت الدراسة برنامجين جديدين لتحسين دقة التنبؤ البنيوي للتفاعلات البروتينية. قام المؤلفون بتطوير DeepMSA2، الذي يستخدم البرمجة الديناميكية المتكررة وخوارزميات نموذج ماركوف المخفية لاستخراج بيانات MSA عالية الجودة بسرعة من مكتبات تسلسل ميتاجينومي ضخمة، ثم يستخدم برنامج DMFold الذي تم تطويره حديثًا لبناء البنية ثلاثية الأبعاد للمجمع البروتيني.
تظهر النتائج التجريبية أن دقة التنبؤ البنيوي لـ DMFold/DeepMSA2 للمجمعات البروتينية أفضل بشكل ملحوظ من دقة الخوارزميات مثل AlphaFold2. على وجه الخصوص، DMFold (https://zhanggroup.org/DMFoldفازت خوارزمية ) بالبطولة في مجال التنبؤ بالبنية المعقدة للبروتين في أحدث مسابقة للتنبؤ بالبنية المعقدة للبروتين (CASP15).

عنوان الورقة:
https://www.nature.com/articles/s41592-023-02130-4
ومؤخرًا، قام الفريق بتوسيع اتجاهه البحثي ليشمل تصميم وتوقع بنية الحمض النووي الريبي والببتيدات القصيرة، واستكشف الموضوعات المتعلقة بتصميم الأدوية. وفي المستقبل، أعتقد أن البروفيسور تشانغ يانغ سيواصل قيادة فريقه لاستكشاف أسرار علم الأحياء.
مراجع:
1.https://www.biorxiv.org/content/10.1101/2025.03.05.641632v1
2.https://mp.weixin.qq.com/s/X_VJ-WOWEP08p5GAJOgq9A








