Command Palette
Search for a command to run...
البروفيسور تشنغ وي من جامعة نانكاي: AlphaFold ليس مثاليًا، ولا يزال لدى المجتمع الأكاديمي الفرصة "للتفوق على المنحنى"

في السنوات الأخيرة، وبمساعدة تقنيات الذكاء الاصطناعي مثل التعلم العميق، تطور مجال التنبؤ ببنية البروتين بسرعة. في أكتوبر 2024، فاز ديميس هاسابيس وجون إم. جامبر من شركة DeepMind بجائزة نوبل في الكيمياء لعام 2024 بفضل AlphaFold. ومع ذلك، هذا لا يعني أن AlphaFold لا يمكن الاستغناء عنه، ولا تزال هناك خوارزميات ممتازة أخرى تستحق الاستكشاف.
في الحلقة السادسة من سلسلة البث المباشر "Meet AI4S"،تشرفت شركة HyperAI بدعوة البروفيسور تشنغ وي، أستاذ في كلية الإحصاء وعلوم البيانات بجامعة نانكاي،تحت عنوان "عرش AlphaFold3 غير مستقر، والمجتمع الأكاديمي يتجاوزه: التنبؤ بالبنية ثلاثية الأبعاد للجزيئات البيولوجية الكبيرة وتفاعلاتها استنادًا إلى التعلم العميق"، شارك الجميع حدود AlphaFold واتجاهات التحسين المستقبلية، بالإضافة إلى الخوارزميات ومواضيع البحث التي تستحق الاستكشاف في المجتمع الأكاديمي.
* قم بمتابعة الحساب الرسمي والرد على "Meet AI4S 6th" للحصول على عرض تقديمي PPT
لقد قامت HyperAI بتنظيم وتلخيص المشاركة المتعمقة دون انتهاك النية الأصلية. وفيما يلي نص الخطاب:
حدود AlphaFold
تشكل البروتينات حجر الزاوية في أنشطة الحياة، ويعد التنبؤ بالهياكل البروتينية ثلاثية الأبعاد أمرًا بالغ الأهمية لفهم الوظائف البيولوجية. على الرغم من أن AlphaFold 2 الذي أطلقته شركة DeepMind قد أخذ التنبؤ ببنية البروتين إلى مستوى جديد، إلا أن هذا لا يعني أن إطار AlphaFold 2 الشامل قد حل جميع مشاكل التنبؤ ببنية البروتين.
أولاً وقبل كل شيء، إذا أخذنا AlphaFold 2 نفسه كمثال، فإنه لا يزال لديه العديد من القيود:
* الدقة تحتاج إلى تحسين
تشير التقارير الرسمية إلى أن AlphaFold 2 يمكنه التنبؤ بالهياكل بدقة تزيد عن 90%، لكن المهمة الفعلية لا يمكنها الوصول إلى هذا المستوى العالي.
* التنبؤ ببنية البروتين متعدد المجالات محدود
يؤدي AlphaFold 2 أداءً جيدًا في التنبؤ بالبروتينات ذات المجال الواحد، ولكن بالنسبة للبروتينات المعقدة متعددة المجالات، حيث تكون المجالات مرنة نسبيًا، فإن دقة التنبؤ ليست جيدة.
* التنبؤ ببنية البروتين المعقدة محدود
تحتاج البروتينات عادة إلى تكوين مجمعات مع بروتينات أخرى لتؤدي وظيفتها، ولكن الإصدار الأولي من AlphaFold 2 لم يعالج هذه المشكلة.
* التنبؤ ببنية الحمض النووي الريبوزي، والتنبؤ ببنية الحمض النووي الريبوزي-الحمض النووي الريبوزي، والتنبؤ ببنية البروتين-الحمض النووي الريبوزي محدودة
وكما ذكرنا أعلاه، لم يتم تناول هذه القضايا في النسخة الأولية.
* التنبؤ بديناميكيات البروتين/التغيرات التكوينية محدود
لا تستطيع طرق التحليل التجريبي عادةً التقاط الحالة البنيوية إلا في لحظة معينة، ولكن البروتينات لا توجد بشكل ثابت في كائناتها الحية، وقد تكون بنيتها في نقاط زمنية مختلفة مختلفة. لم يتم حل هذه المشاكل بعد بواسطة AlphaFold 2.
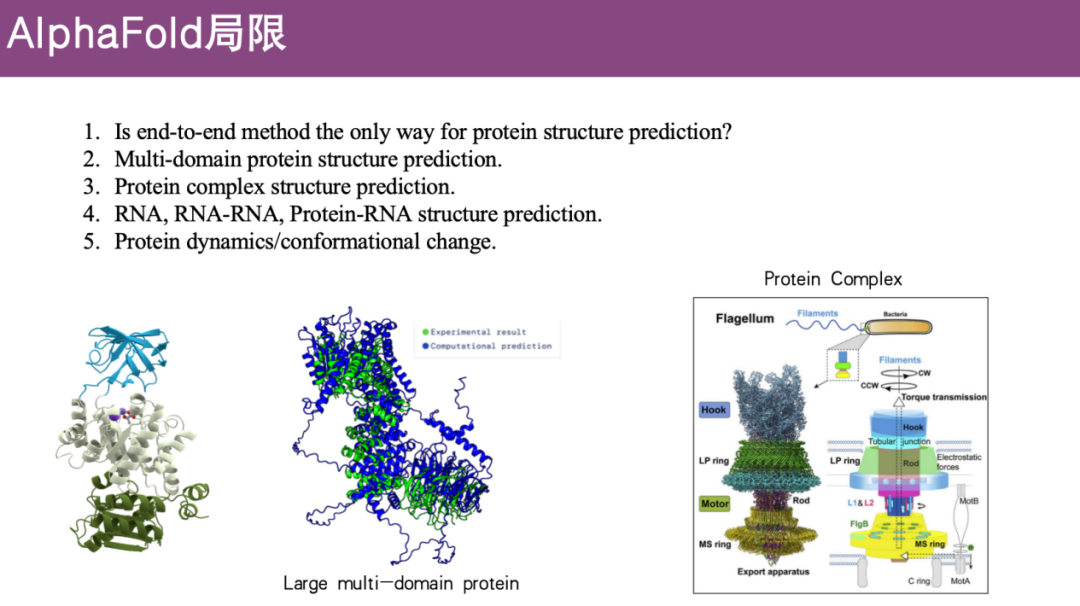
وعلاوة على ذلك، على الرغم من أن DeepMind قامت بتكرار AlphaFold 3، ونحن جميعًا نعلم أنه يعمل بشكل جيد في التنبؤ بهياكل مونومر البروتين، إلا أن دقته في التنبؤ بالمجمعات والأحماض النووية والجزيئات الصغيرة لا تزال بحاجة إلى التحسين. لذلك،قد يضيف الجيل القادم من AlphaFold وحدات تنبؤ ذات وظائف أخرى.على سبيل المثال، نظرًا لأن النماذج الحالية تُستخدم بشكل أساسي للتعامل مع الهياكل الثابتة، فسوف نستكشف العمليات الديناميكية الجزيئية ونتنبأ بتكوينات البروتين. بالإضافة إلى ذلك، قد يشمل هذا أيضًا مجال تصميم البروتين، مما يعكس عملية التنبؤ بأكملها.
لذلك، حتى مع AlphaFold، لا يزال هناك الكثير من العمل الذي يتعين القيام به في جميع أنحاء الأوساط الأكاديمية.
بالإضافة إلى AlphaFold، هل هناك أي طرق أخرى تستحق الاستكشاف؟
في الماضي، كانت الطرق الرئيسية التي استخدمناها لحل البنية الثلاثية الأبعاد للبروتينات هي الأشعة السينية، والرنين المغناطيسي النووي (NMR)، والمجهر الإلكتروني المبرد. ونظراً لصعوبة وارتفاع تكلفة حل هياكل البروتين تجريبياً، فقد تحتاج بعض الفرق إلى قضاء أشهر أو سنوات لحل البنية الثلاثية الأبعاد للبروتين. ونتيجة لذلك، بدأ الناس في استكشاف طريقة أكثر اقتصادا وسرعة، وهي التنبؤ ببنية البروتين من خلال الخوارزميات.
نحن نعلم أن البروتينات تتكون بشكل أساسي من 20 نوعًا من الأحماض الأمينية، والتي يتم تمثيلها عادةً بالأحرف الإنجليزية، وتحتوي جزيئات الأحماض الأمينية أيضًا على العديد من الذرات.لذلك، يمكن تلخيص مشكلة التنبؤ ببنية البروتين على النحو التالي: إدخال سلسلة من الأحماض الأمينية تتكون من هذه الأحرف، واستخدام خوارزمية حسابية للتنبؤ بالإحداثيات المكانية ثلاثية الأبعاد (x، y، z) لكل ذرة في كل حمض أميني في تسلسل البروتين.
عند النظر إلى تاريخ تطور التنبؤ ببنية البروتين بالكامل، فقد ظهرت مجموعة متنوعة من الخوارزميات التمثيلية في مراحل مختلفة، مثل النمذجة المقارنة أو نمذجة التماثل، ومحاكاة الديناميكيات الجزيئية (MD)، وخوارزمية الترابط، والتنبؤ الجديد، وخوارزمية التنبؤ بالبنية القائمة على التنبؤ بالتعلم العميق لخرائط الاتصال. المقدمة الرئيسية هي كما يلي:
* النمذجة المقارنة أو النمذجة المتجانسة
تعتمد هذه الطريقة على مبادئ التطور البيولوجي.ويعتقد أنه إذا كان التشابه التسلسلي مرتفعًا، فإن بنية ووظيفة البروتين ستكون أيضًا متشابهة نسبيًا.لذلك، يمكننا أولاً الحصول على تسلسل الأحماض الأمينية للبروتين غير المعروف، ثم العثور على قالب بنية البروتين المحلولة مع تشابه التسلسل العالي في قاعدة بيانات PDB من خلال محاذاة التسلسل، والتنبؤ ببنية البروتين غير المعروف من خلال الهجرة أو المحاذاة.
*تحتوي قاعدة بيانات PDB على هياكل البروتينات التي تم حلها في هذا المجال

* محاكاة الديناميكيات الجزيئية
الفكرة الأساسية هي إنشاء بنية ثلاثية الأبعاد أولية بشكل عشوائي بناءً على تسلسل الأحماض الأمينية للبروتين، وتعيين إحداثيات عشوائية لكل ذرة، وضبط الموضع الذري، ثم حساب طاقة حالة البروتين في أوقات مختلفة بناءً على مجال الطاقة الفيزيائي الذي تم إنشاؤه مسبقًا.البنية التي تحتوي على أقل قدر من الطاقة هي التركيب البروتيني المعقول.

* خوارزمية الترابط
على غرار نمذجة التماثل، فإن الفرق هو أنه على الرغم من أن البروتينات ذات التشابه العالي في التسلسل غالبًا ما تكون متشابهة في البنية، إلا أن البروتينات ذات البنيات المتشابهة قد يكون لها تشابه منخفض في التسلسل، ولا يمكن لهذه البروتينات العثور على معلومات قالب مناسبة في قاعدة بيانات PDB. ثم اقترح الباحثون مفهوم الملف الشخصي، واستنادًا إلى التسلسلات المتجانسة التي تم جمعها، استخدموا محاذاة التسلسلات المتعددة (MSA) لمواءمة الأحماض الأمينية المختلفة بنفس طريقة محاذاة ملفين بروتينيين.
وهذا يعني أنه حتى لو كانت تسلسلات الأحماض الأمينية مختلفة،لكن ملامحهم متشابهة، لذا يمكننا أن نفترض أن بنيتهم متشابهة.استخدم هذا للعثور على القوالب.

* التنبؤ الجديد
قد لا تحتوي بعض البروتينات على هياكل مماثلة في قاعدة البيانات.حاول الباحثون بعد ذلك إجراء تنبؤات من خلال تقسيم تسلسل البروتين بأكمله إلى أجزاء أقصر، والبحث عن قوالب لهذه الأجزاء الصغيرة في قاعدة البيانات، ثم تجميع هذه القوالب الصغيرة في بنية ثلاثية الأبعاد كاملة.
على وجه التحديد، قام البروفيسور ديفيد بيكر من جامعة واشنطن بتطوير برنامج روزيتا، الذي يتمثل مبدأه الرئيسي في تفكيك تسلسل البروتين إلى العديد من الأجزاء الصغيرة، وتجميع هذه الأجزاء عشوائيًا، ثم تحسينها باستخدام دالة الطاقة التي تم تطويرها في محاكاة الديناميكيات الجزيئية، وإجراء التنبؤ بالبنية من خلال مبادئ مماثلة للمحاكاة الديناميكية وتقليل الطاقة.
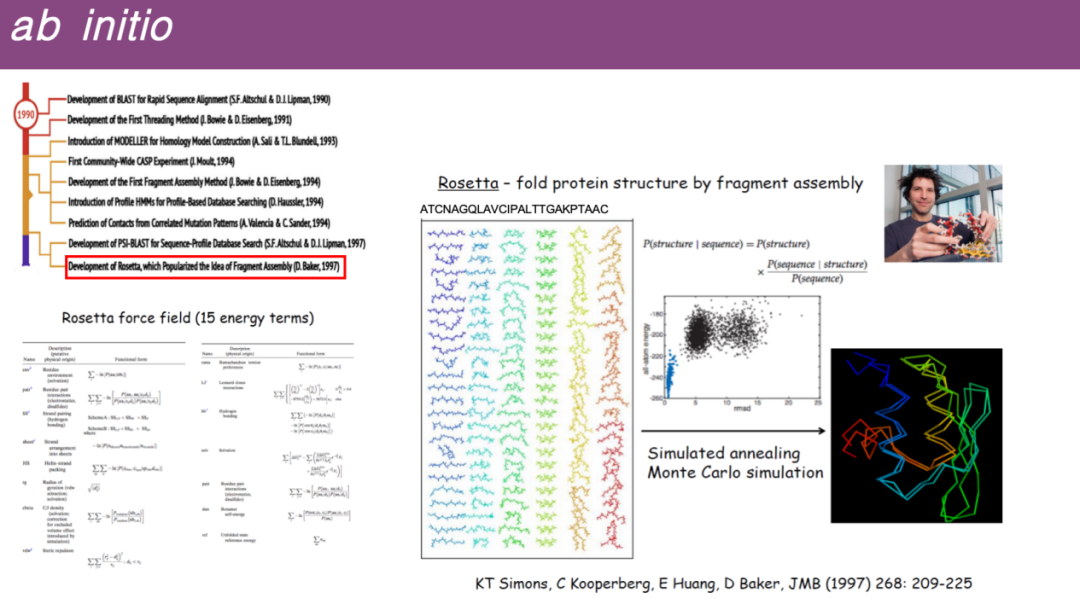
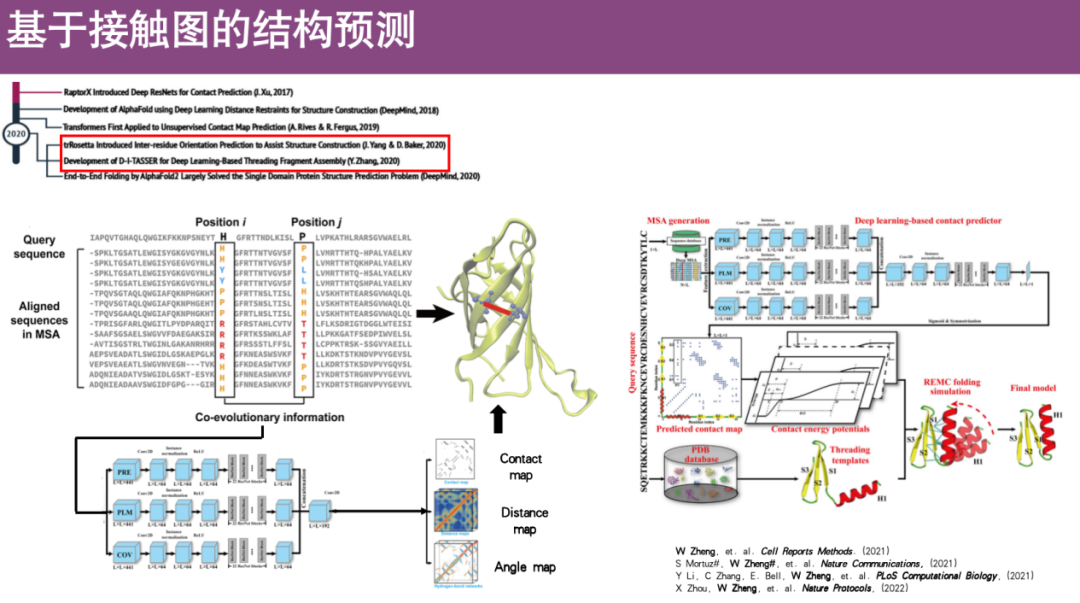
* خريطة الاتصال
الفكرة الرئيسية هي تحويل البنية الثلاثية الأبعاد للبروتين إلى رسم بياني ثنائي الأبعاد.يتم استخدام معلومات البنية ثلاثية الأبعاد للبروتين، أي يتم استخدام مواضع إحداثيات جميع النقاط المكانية لحساب المسافة بين الأحماض الأمينية المختلفة. من المفترض أن الاتصال يتشكل عندما تكون المسافة بين حمضين أمينيين أقل من حد معين، وإلا فلن يتشكل أي اتصال. يتم استخدام هذا التعريف لضغط البنية ثلاثية الأبعاد في رسم بياني ثنائي الأبعاد. علاوة على ذلك، يمكن استخدام معلومات خريطة الاتصال ثنائية الأبعاد هذه لإعادة بناء البنية ثلاثية الأبعاد للبروتين.
وعلى وجه التحديد، قام الباحثون بتطوير العديد من الأساليب المبنية على التعلم العميق. الفكرة الأساسية هي بناء محاذاة تسلسل متعددة (MSA) أولاً لمراقبة المعلومات التطورية المشتركة للملفات الشخصية في الأحماض الأمينية i وj، لأن مثل هذه الأحماض الأمينية المتطورة المشتركة غالبًا ما تكون قريبة جدًا في الفضاء وستشكل اتصالات. بعد ذلك، يتم إدخال معلومات التطور المشترك كميزات في شبكة التعلم العميق للتدريب، وبالتالي التنبؤ بخريطة اتصال البروتين واستعادة بنية البروتين ثلاثية الأبعاد.
على سبيل المثال، قام فريق البروفيسور تشنغ وي في السابق بتطوير خوارزمية تسمى CI-TASSER، وهي حاليًا طريقة شائعة الاستخدام للتنبؤ ببنية البروتين استنادًا إلى خرائط الاتصال.
أخيرًا، يدمج AlphaFold المبادئ الأساسية للعديد من الخوارزميات المذكورة أعلاه ويبني بنجاح إطار عمل متكامل يمكنه إدخال تسلسلات البروتين بشكل مباشر ثم إخراج الهياكل.
من خلال أخذ إنجازات الفريق كمثال، واستكشاف الفرص المتاحة للتفوق على الأكاديميين
إن التنبؤ ببنية البروتين له تأثير كبير على المجال الطبي الحيوي، على سبيل المثال،تتضمن الخوارزميات التي طورها فريق البروفيسور تشنغ وي حاليًا التنبؤ بالهياكل البروتينية الفيروسية غير المعروفة (فيروس كورونا الجديد)، والمساعدة في تحليل الهياكل البروتينية باستخدام المجهر الإلكتروني المبرد، ومساعدة علماء الأحياء على فهم الوظائف التطورية للبروتين، وفحص الأجسام المضادة.
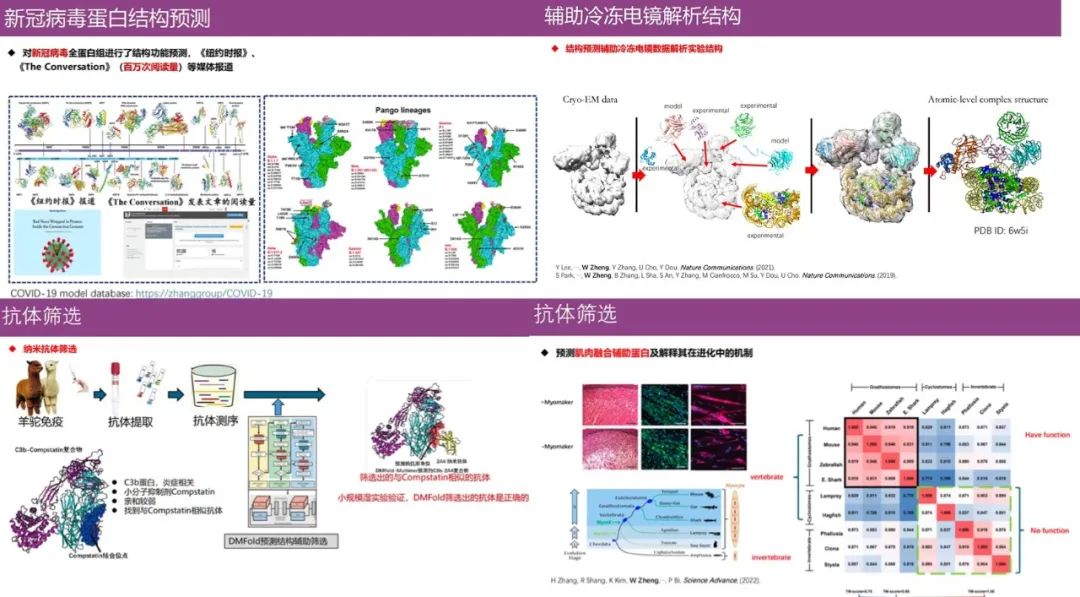
وبالإضافة إلى ذلك، وكما هو موضح في الشكل أدناه، تم تحويل جميع خوارزميات التنبؤ بمونومر البروتين والبنية المعقدة التي طورها الفريق إلى خوارزميات خادم أوتوماتيكية ونشرها على موقع مجموعة البحث. وقد خدمت خوارزمياتها أكثر من 90 ألف مستخدم في أكثر من 100 دولة حول العالم، ويمكن للجميع استخدامها.
*عنوان المشروع الإجمالي:
https://seq2fun.dcmb.med.umich.edu/DMFold

طريقة التنبؤ ببنية مونومر البروتين DI-TASSER
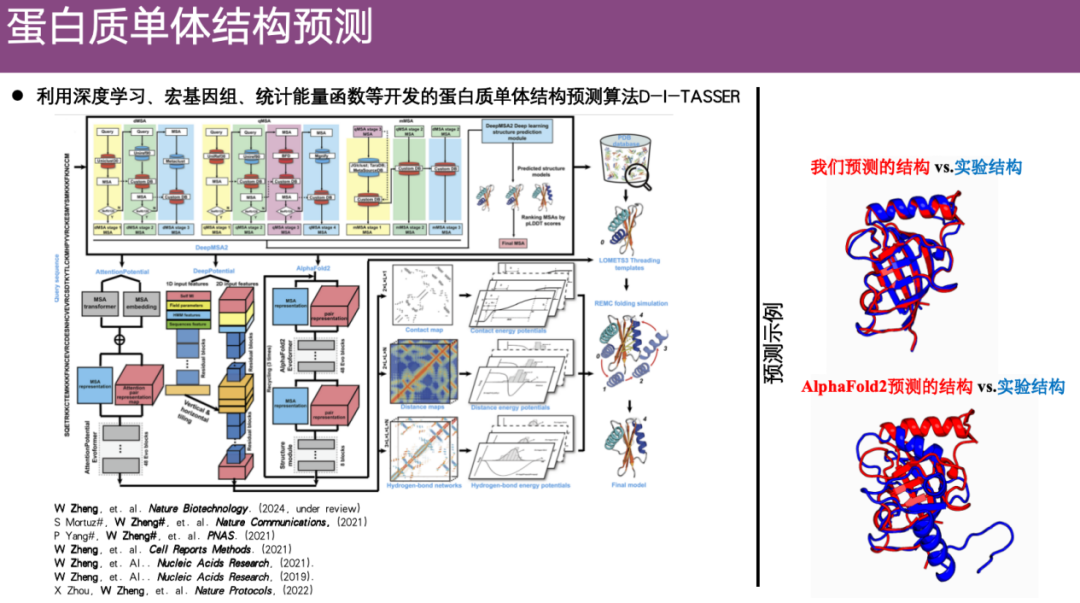
لقد جذبت مشكلة التنبؤ ببنية مونومر البروتين الكثير من الاهتمام دائمًا. قبل AlphaFold 2، كان فريق البروفيسور Zheng Wei يجري أبحاثًا للتنبؤ بالبنية استنادًا إلى خرائط الاتصال. بعد ظهور AlphaFold 2، بدأ الفريق يتساءل عما إذا كان بإمكانهم دمج القيود المكانية مثل خريطة الاتصال التي تنبأ بها AlphaFold 2 في الخوارزميات التي تم تطويرها مسبقًا. لذلك، بناءً على القيود المكانية، والميتاجينومات، ووظائف الطاقة الإحصائية، وما إلى ذلك.قام الفريق بتطوير خوارزمية التنبؤ ببنية المونومر البروتيني DI-TASSER، والتي أظهرت نتائج جيدة بعد التحسين.
كما هو موضح في المثال الموجود على الجانب الأيمن من الشكل أدناه، يمثل اللون الأحمر بنية البروتين التي تنبأت بها DI-TASSER، ويمثل اللون الأزرق البنية التي تم تحليلها تجريبياً. كما ترون،إن البنية التي تنبأت بها DI-TASSER تشبه إلى حد كبير البنية التي تم حلها تجريبياً.وعلى النقيض من ذلك، فإن البنية التي تنبأ بها AlphaFold 2 تختلف بشكل كبير عن البنية التجريبية حتى بعد المحاذاة، كما أن دقة التنبؤ بها أقل قليلاً.
بالإضافة إلى ذلك، يتم تقييمه على مجموعات بيانات البروتين المتعددة. كما هو موضح في الجانب الأيمن من الشكل أدناه، عند التنبؤ بمجال واحد ومجالات متعددة،دقة التنبؤ الخاصة بـ DI-TASSER أعلى من دقة AlphaFold 2 وحتى أعلى من دقة AlphaFold 3.
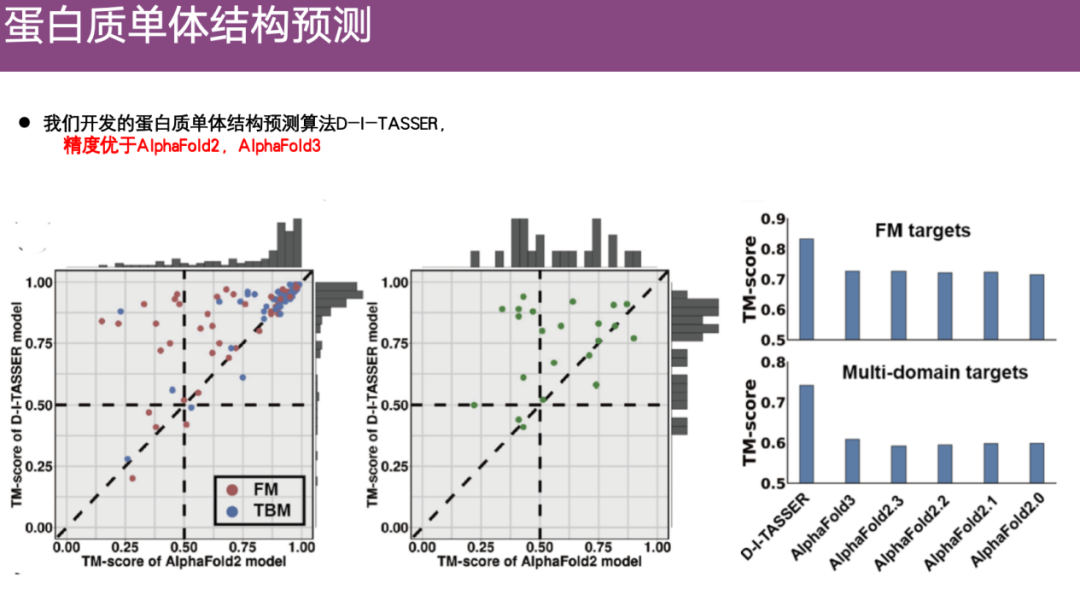
ولضمان مصداقية التقييم، لم يقم الفريق بإجراء تقييمات داخلية فحسب، بل شارك أيضًا في المنافسة الموثوقة في هذا المجال - CASP.
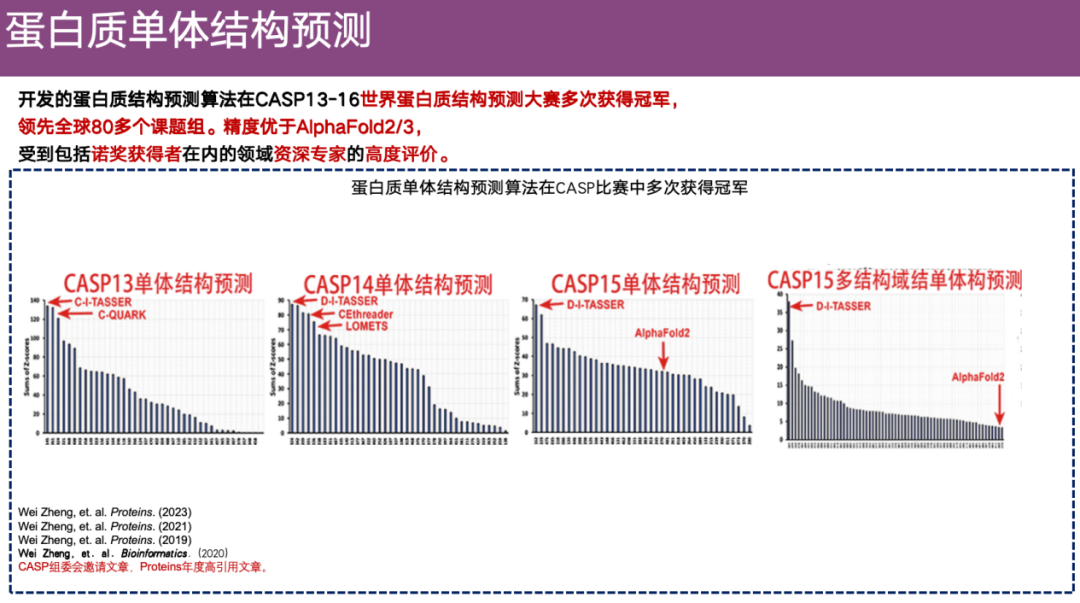
تُعرف مسابقة CASP باسم الألعاب الأولمبية في هذا المجال وتهدف في المقام الأول إلى توحيد أساليب التقييم للتنبؤ ببنية البروتين. نظرًا لوجود العديد من أنواع خوارزميات التنبؤ ببنية البروتين ثلاثية الأبعاد، فقد قام كل مختبر أيضًا بتطوير خوارزميته الخاصة. نظرًا لأن مجموعات بيانات التقييم والأساليب قد تختلف، فإن كل مجموعة بحثية تدعي عادةً أن أسلوبها هو الأكثر دقة في العالم. ولحل هذه الفوضى، تم إنشاء مسابقة CASP.
اعتبارًا من العام الماضي، تم عقد CASP بنجاح لمدة 16 جلسة واستمر لمدة 32 عامًا، وجذب العديد من الفرق الموثوقة للمشاركة، مثل فريق البروفيسور ديفيد بيكر وفريق DeepMind.
شاركت DI-TASSER وخوارزمياتها السابقة في مسابقات CASP عدة مرات. خلال CASP 13-CASP 15، كانت هذه الطريقة في موقع الريادة في مجال التنبؤ ببنية مونومر البروتين. في CASP 15،كما شاركت خوارزمية DI-TASSER أيضًا في التقييم متعدد المجالات، وكانت دقتها الإجمالية أفضل من دقة جميع مجموعات البحث المشاركة.
DMFold، طريقة التنبؤ ببنية البروتين المعقدة
التحدي الرئيسي في التنبؤ بالبنية المعقدة هو التنبؤ بالالتواء النسبي بين بروتينين، والذي يمكن تحليله باستخدام المعلومات التطورية المشتركة.
على سبيل المثال، من خلال بناء محاذاة تسلسل متعددة (MSA) من وحدات البروتين، ودمج تسلسلي MSA في تسلسل واحد استنادًا إلى بعض طرق الاتصال، واستخدام العلاقة التطورية المشتركة للأحماض الأمينية بين تسلسلي MSA لاستنتاج المسافة بين الأحماض الأمينية في البروتينات المختلفة، يمكن أيضًا دمج المعلومات التطورية المشتركة في إطار التعلم العميق للتنبؤ بالالتواء النسبي بين بروتينين.
في هذا الصدد،قامت مجموعة البحث التابعة للبروفيسور تشنغ وي بتطوير خوارزميات DeepMSA و MetaSource لبناء محاذاة تسلسل متعددة أعمق.بالإضافة إلى ذلك، استخدم الفريق أيضًا التعلم العميق والميتاجينوميات وما إلى ذلك لتطوير خوارزمية التنبؤ بالبنية المعقدة للبروتين DMFold.
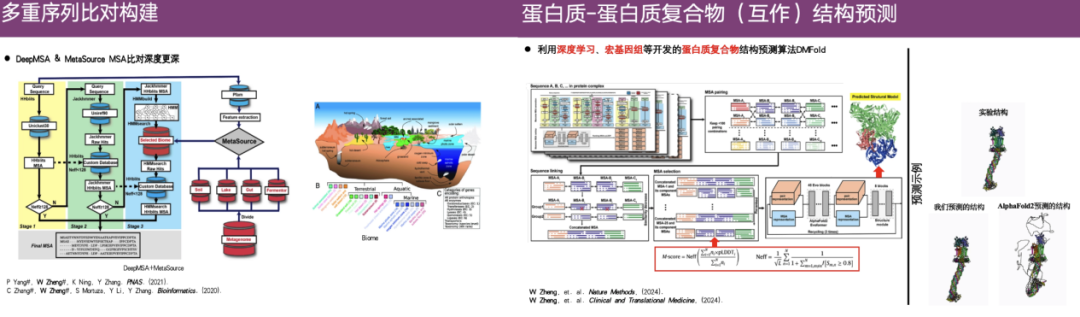
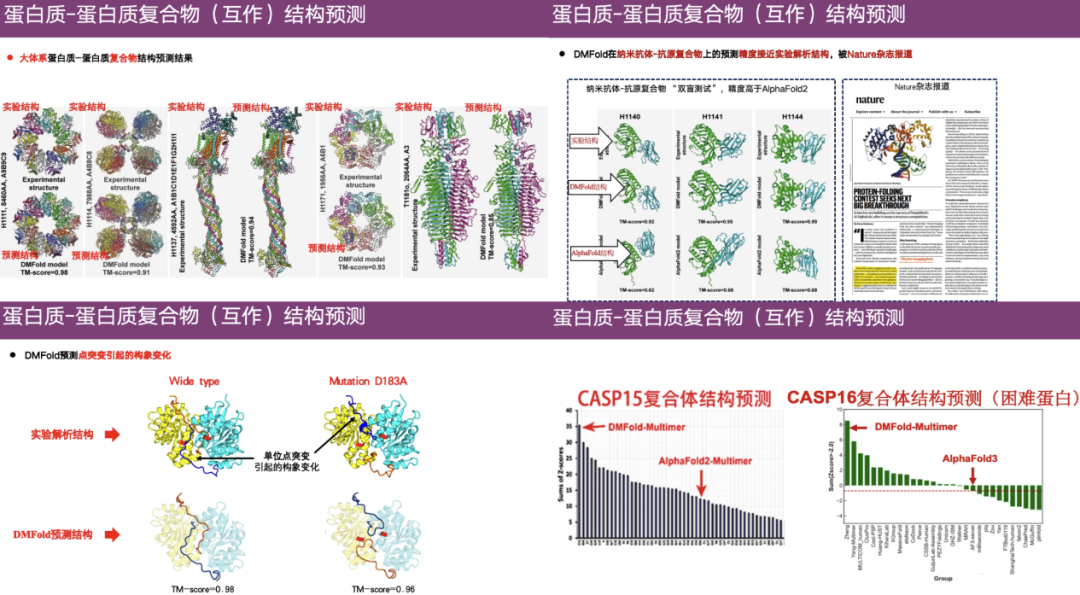
كما هو موضح في الحالة الموجودة في أقصى يمين الشكل أعلاه، فإن الجزء العلوي هو البنية الحقيقية التي تم الحصول عليها من خلال التحليل التجريبي، والجزء السفلي الأيسر هو البنية التي تنبأت بها DMFold، والجزء الأيمن هو النتيجة التي تنبأت بها AlphaFold 2. يمكن ملاحظة أن البنية التي تنبأت بها AlphaFold 2 فوضوية نسبيًا ولها امتدادات غير طبيعية تشبه المجسات. وعلى النقيض من ذلك، فإن البنية المتوقعة لـ DMFold تشبه إلى حد كبير البنية التجريبية.يوضح هذا أن خوارزمية DMFold متفوقة على AlphaFold 2 في التنبؤ بالبنية المعقدة.
بالإضافة إلى ذلك، يُظهر DMFold أيضًا دقة عالية في مجمعات البروتين-البروتين ذات النظام الكبير، ومجمعات الأجسام المضادة النانوية-المستضد، والتغيرات التكوينية الناجمة عن الطفرات النقطية، وما إلى ذلك. في مسابقة CASP 15، إن التصنيف الإجمالي لـ DMFold أعلى بكثير من AlphaFold 2، وفي CASP 16، يعتبر DMFold أيضًا أفضل من AlphaFold 3.
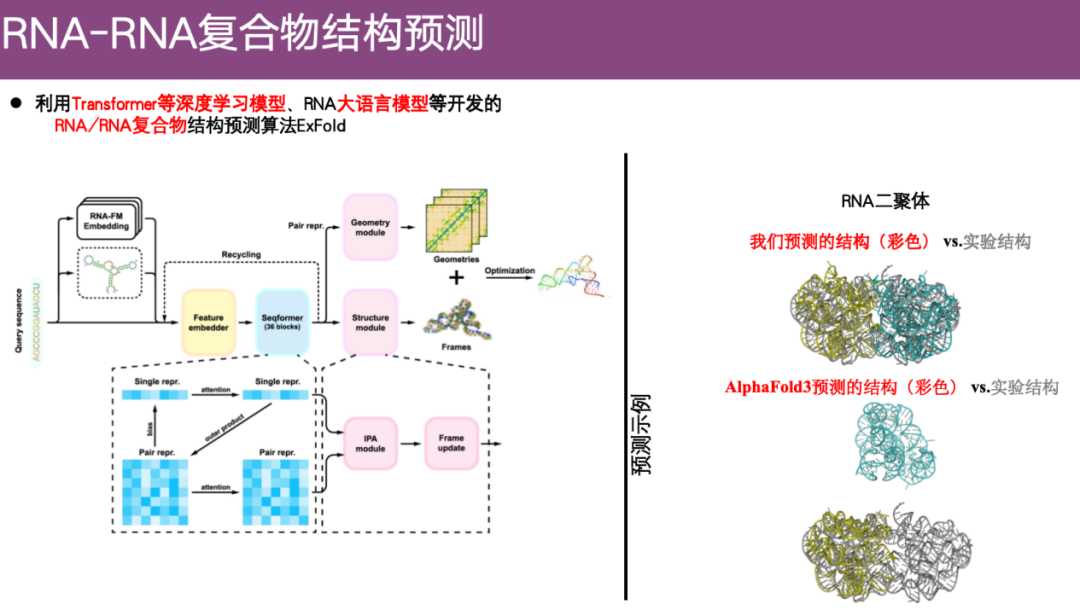
طريقة التنبؤ ببنية مجمع RNA-RNA ExFold
وفي السنوات الأخيرة، بدأ الفريق بالتركيز على مشكلة التنبؤ ببنية الحمض النووي الريبي. على سبيل المثال، قاموا بتطوير خوارزمية التنبؤ بالبنية المعقدة RNA/RNA ExFold باستخدام نماذج التعلم العميق مثل Transformer ونماذج اللغة الكبيرة RNA.
كما هو موضح في المثال الموجود على الجانب الأيمن من الشكل أدناه، فإن الجزء الرمادي هو الهيكل التجريبي والجزء الملون هو الهيكل المتوقع. كما ترون،وباستخدام طريقة ExFold، تم محاذاة الهيكلين بشكل جيد. في المقابل، أظهر تنبؤ AlphaFold 3 أنه لم يكن هناك حتى أي اتصال بين جزيئي الحمض النووي الريبي، وهو ما يمكن اعتباره خاطئًا تمامًا تقريبًا.
كما قام الفريق أيضًا بمقارنة دقة ExFold 3 مع AlphaFold 3 باستخدام مجموعة بيانات أكبر، كما هو موضح على الجانب الأيسر من الشكل أدناه. يمثل المحور Y دقة التنبؤ لـ ExFold.يمثل المحور X دقة التنبؤ الخاصة بـ AlphaFold 3. ويمكن ملاحظة أن مزايا ExFold لا تزال واضحة تمامًا.
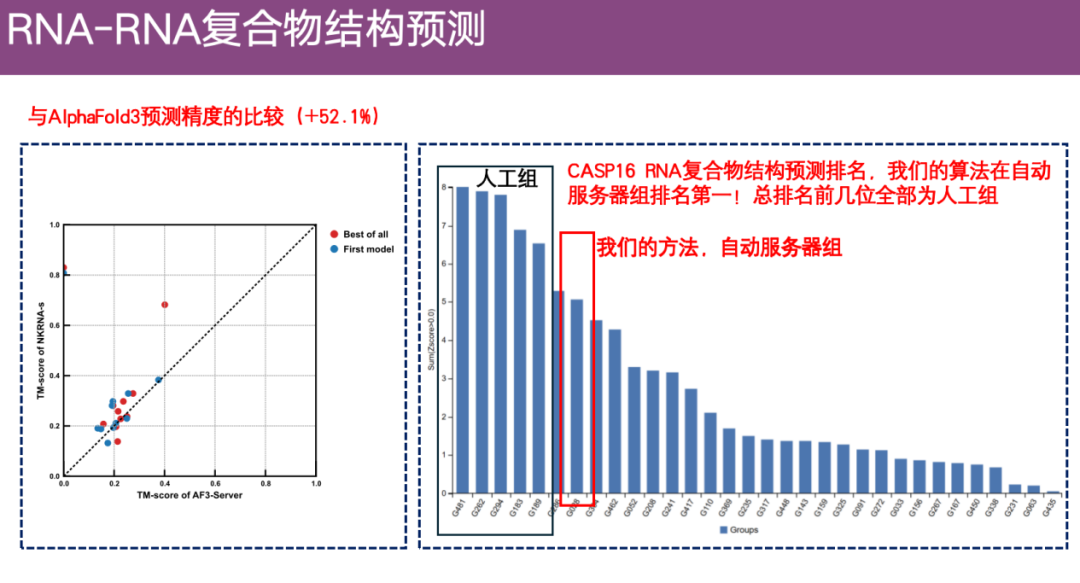
بالإضافة إلى ذلك، في مسابقة التنبؤ ببنية مجمع RNA CASP 16،على الرغم من أن ExFold لا يحتل المرتبة الأولى بشكل عام، إلا أنه يحتل المرتبة الأعلى بين جميع الخوارزميات التلقائية (خوارزميات الخادم).
* تنقسم مسابقة CASP إلى مجموعة آلية ومجموعة يدوية. يجب على المجموعة التلقائية تقديم نتائج التنبؤ بشكل تلقائي بالكامل خلال 3 أيام، ولا يُسمح بالتدخل البشري؛ المجموعة اليدوية لها 3 أسابيع، ويُسمح لها بإضافة خبرة الخبراء والتعديلات اليدوية.
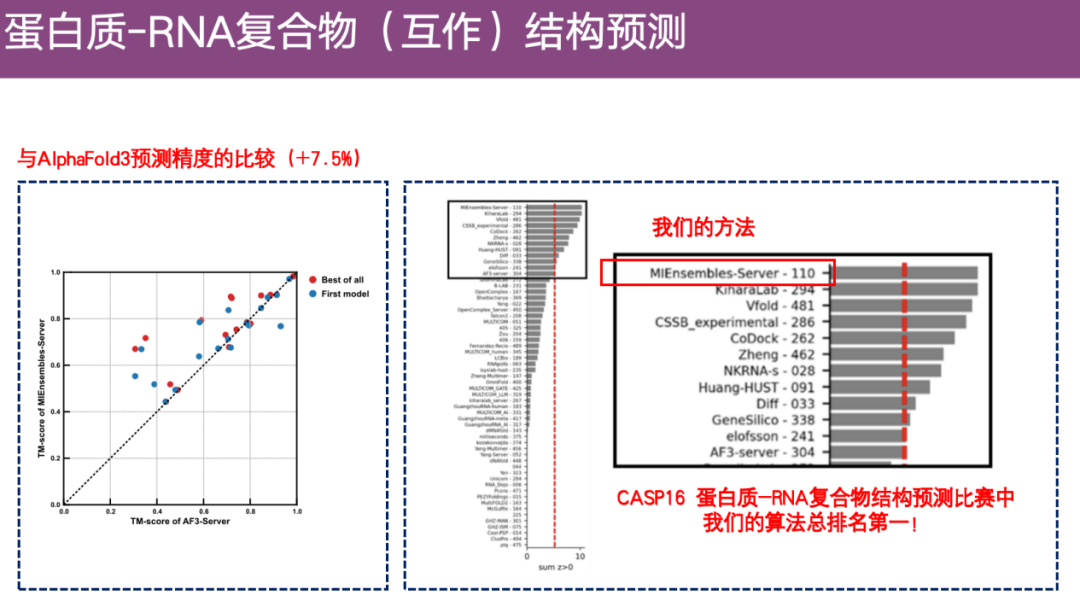
طريقة التنبؤ ببنية مجمع البروتين-RNA DeepProtNA
وفيما يتعلق بمشكلة التنبؤ ببنية مجمع البروتين-RNA، استخدم الفريق نماذج التعلم العميق مثل Transformer ونموذج اللغة الكبيرة الشائع مؤخرًا لتطوير خوارزمية جديدة للتنبؤ بالبنية - DeepProtNA.
كما هو موضح في المثال الموجود على اليمين أدناه، في مجمع الأجسام المضادة-RNA، تمثل الألوان نتائج التنبؤ الخاصة بـ DeepProtNA، بينما يمثل اللون الرمادي البنية التجريبية. بعد المحاذاة، يمكننا أن نجد أنإن البنية المتوقعة لـ DeepProtNA متوافقة إلى حد كبير مع البنية التجريبية (تداخل اللون الرمادي واللون).وخاصةً عند الواجهة بين بروتين الجسم المضاد وRNA للمستضد، فإن دقة التنبؤ عالية جدًا. على النقيض من ذلك، فإن البنية المتوقعة لـ AlphaFold 3 لا تتداخل مع البنية التجريبية، وتأثير التنبؤ ضعيف.
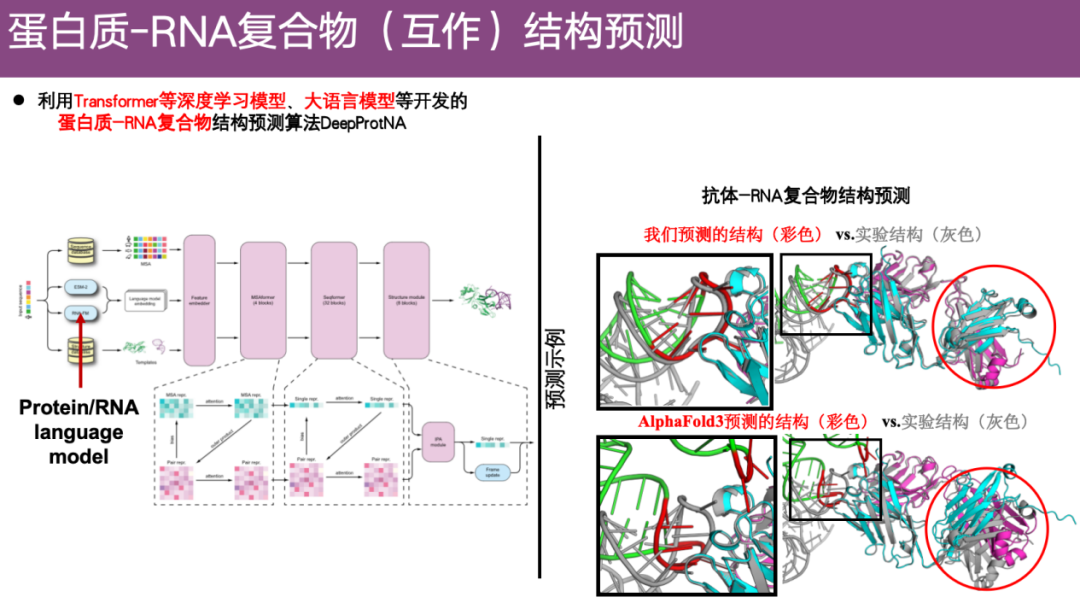
أيضًا،يعتبر DeepProtNA أكثر دقة من AlphaFold 3 بنحو 7.5 نقطة مئوية.حصل على المركز الأول في مسابقة مجموعة الخوادم CASP 16.
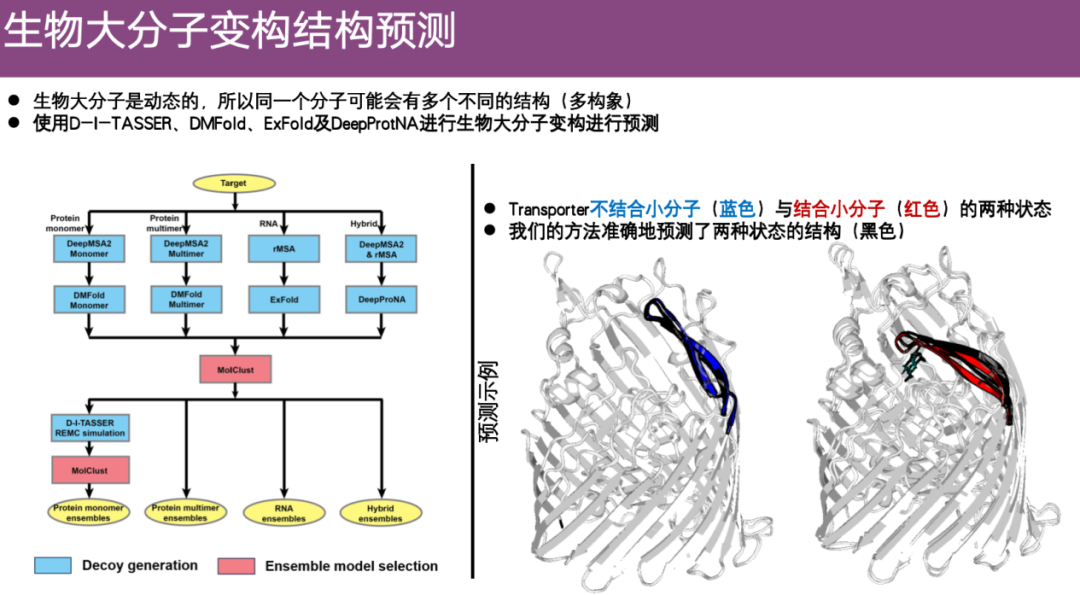
EnsembleFold: طريقة للتنبؤ بالهياكل غير المتجانسة للجزيئات الحيوية الكبيرة
ويركز الفريق أيضًا على مشكلة التنبؤ بالبنية غير المتجانسة للجزيئات البيولوجية الكبيرة. إن مدخلات مشكلة التكوين المتعدد الجزيئات هي تسلسل بروتيني، والمخرجات هي إطارات رئيسية متعددة للبروتين في حالات مختلفة. وهذا يعني أنه بالمقارنة مع خوارزميات التنبؤ الثابتة، هناك حاجة إلى التنبؤ بهياكل متعددة مختلفة من تسلسل واحد من الأحماض الأمينية، وتمثل هذه الهياكل الإطارات الرئيسية للعملية الديناميكية بأكملها. هذا موضوع حظي باهتمام كبير في المجال الحالي ولكن من الصعب التنبؤ به.
من خلال دمج الأساليب التي تم تطويرها مسبقًا وتحسينها للتفاعلات الجزيئية الكبيرة،قام فريق البحث بتطوير بعض خوارزميات التجميع وتمكن في النهاية من تشكيل خوارزمية تسمى EnsembleFold.
كما هو موضح في المثال الموجود على الجانب الأيمن من الشكل أدناه، يتم توضيح التغيرات التكوينية للبروتين بعد الارتباط بالجزيء الصغير. يمثل اللون الأزرق البنية التجريبية عندما لا يكون الجزيء الصغير مرتبطًا، ويمثل اللون الأحمر الميل والتغيير التكويني بعد الارتباط بالجزيء الصغير الأخضر. وتوقع الفريق وجود بنيتين بناءً على تسلسل البروتين المدخل، وهما الأجزاء السوداء. يمكن ملاحظة أن البنية المتوقعة لـ EnsembleFold متوافقة جدًا مع البنية الفعلية عندما لا تكون مرتبطة بجزيئات صغيرة. بعد الارتباط بالجزيئات الصغيرة، يمكن لـ EnsembleFold أيضًا أن يتناسب مع البنية التجريبية بشكل جيد. لذلك،يُظهر EnsembleFold دقة عالية للغاية في التنبؤ بالتغيرات التكوينية في الجزيئات الحيوية الكبيرة.
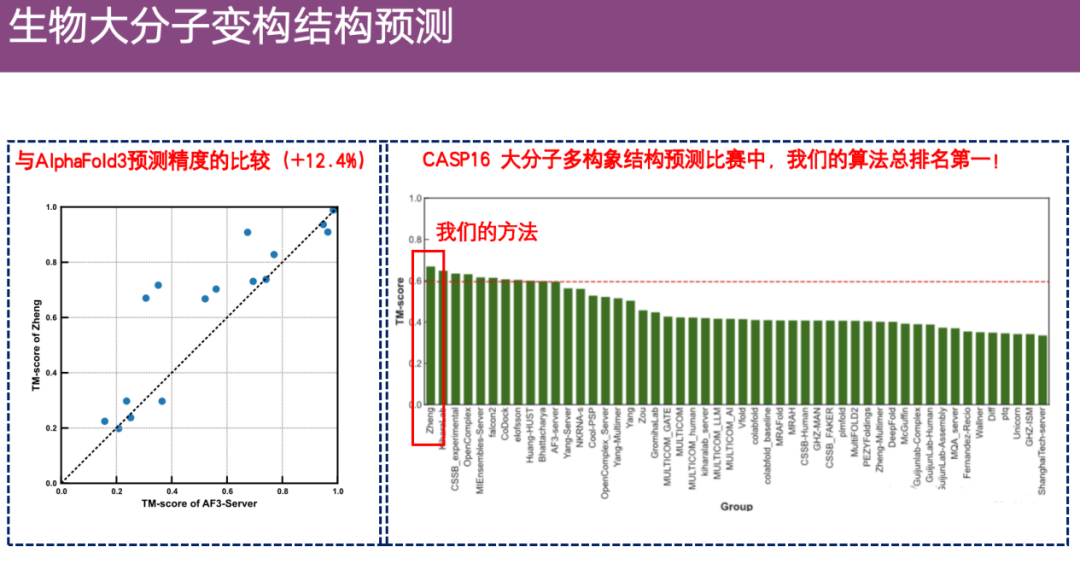
في نفس الوقت،بعد المقارنة مع AlphaFold 3، وجد أن دقة EnsembleFold كانت أعلى بحوالي 12.4%.يحتل المرتبة الأولى بين جميع مسابقات تكوين الجزيئات الكبيرة في CASP 16.
ومن الأمثلة المثيرة للاهتمام هو تنبؤ الفريق بالتغيرات التكوينية لإنزيم تكامل الحمض النووي للبكتيريا في CASP. كما هو موضح في الشكل أدناه، يتم تمثيل تسلسل الأحماض الأمينية للبكتيريا بواسطة P-P'، ويتم تمثيل تسلسل المادة الوراثية للبكتيريا بواسطة B-B'. من خلال عملية ديناميكية، يقوم إنزيم إنتيغريز الحمض النووي للبكتيريا بدمج المادة الوراثية P' للبكتيريا في المادة الوراثية B للبكتيريا لتشكيل B-P'، ويتغير التكوين.
استخدم الفريق خوارزميات للتنبؤ بهذه التغيرات التكوينية المتعددة. تظهر الهياكل التجريبية على اليسار، مع الحالة غير المتكاملة (التكوين 1) في الأعلى والحالة المتكاملة (التكوين 2) في الأسفل. ومن الممكن ملاحظة أن توقعات فريق البحث يمكن أن تعكس بدقة هذين التكوينين المختلفين.
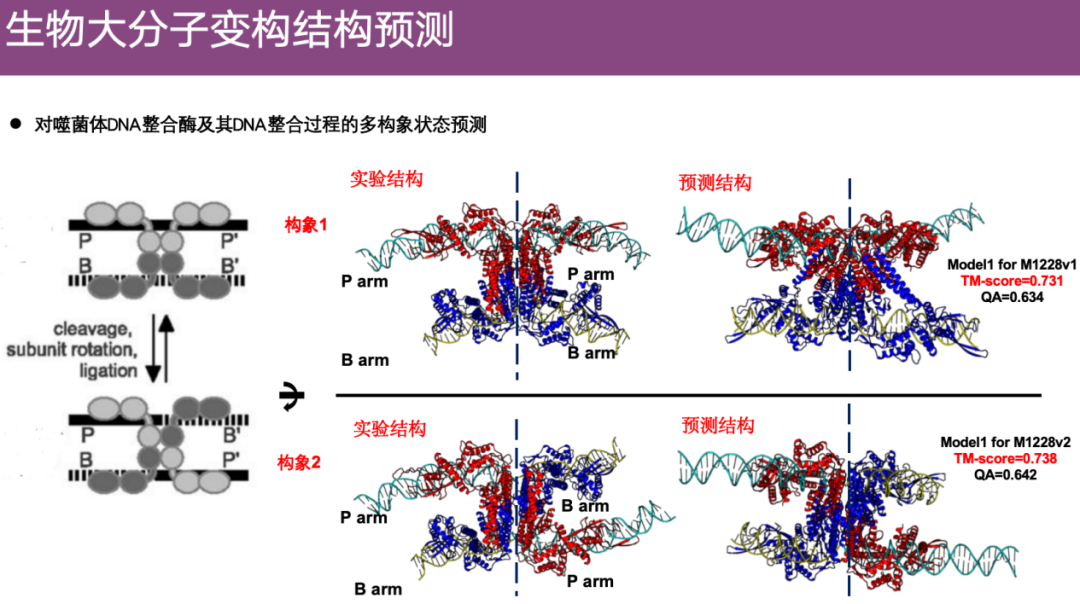
ومن الجدير بالذكر أنه في مسابقة CASP 16،لم يتلق المتسابقون سوى معلومات التسلسل ولم يعرفوا العملية البيولوجية المحددة أو تفاصيل التغيرات التكوينية. ومع ذلك، نجح فريق البروفيسور تشنغ وي في استعادة العملية البيولوجية بأكملها من خلال التنبؤ.وفي أثناء تلخيص المباراة، أعرب الحكام أيضًا عن دهشتهم.
توظيف مجموعة البحث
لقد كان البروفيسور تشنغ وي من كلية الإحصاء وعلوم البيانات في جامعة نانكاي ملتزمًا منذ فترة طويلة بأبحاث التنبؤ ببنية ووظيفة وتفاعل الجزيئات البيولوجية الكبيرة مثل البروتينات. لقد قاد تطوير عدد من خوارزميات التنبؤ ببنية مجمع البروتين والبروتين المعقد والأحماض النووية والمعقدة وخوارزميات تقييم البنية بدقة أفضل من AlphaFold2 / 3. فاز بالبطولة في العديد من مسابقات مسابقة التنبؤ ببنية البروتين العالمية (CASP) (CASP13-16)، وقاد أكثر من 80 مجموعة بحثية أكاديمية/صناعية حول العالم.
ويقوم فريق المعلوماتية الحيوية في كلية الإحصاء وعلوم البيانات بجامعة نانكاي حيث يعمل بتجنيد أعضاء جدد.إذا كنت مهتمًا بعلم الأحياء البنيوي الحسابي، أو المعلوماتية الحيوية، أو علم البيانات، سواء كنت طالب ماجستير أو دكتوراه أو باحث ما بعد الدكتوراه، فنحن نرحب بك للانضمام إلى فريق البروفيسور تشنغ وي.
يمكن للطلاب المهتمين الاتصال بالبروفيسور تشنغ وي عبر الطرق التالية:
* البريد الإلكتروني: [email protected]
* وي تشات: 18622152765









