Command Palette
Search for a command to run...
أشار ليكون إلى أن جامعة كاليفورنيا في بيركلي وآخرون. اقترح طريقة توليد البروتين متعدد الوسائط PLAID، والتي تولد تسلسلات وهياكل بروتينية كاملة الذرات في نفس الوقت

على مدى السنوات الماضية، واصل العلماء استكشاف بنية وتكوين البروتينات من أجل كشف "شفرة الحياة" بشكل أفضل.يتم تحديد وظيفة البروتين من خلال بنيته، بما في ذلك هوية وموقع ذرات السلسلة الجانبية والرئيسية وخصائصها البيوفيزيائية، والتي يشار إليها بشكل جماعي باسم البنية الذرية الشاملة.ومع ذلك، لتحديد مكان وضع ذرات السلسلة الجانبية، يجب علينا أولاً معرفة التسلسل. لذلك، يمكن النظر إلى توليد بنية الذرات الكاملة باعتبارها مشكلة متعددة الوسائط تتطلب التوليد المتزامن للتسلسل والبنية.
ومع ذلك، فإن طرق بناء بنية البروتين وتوليد التسلسل الموجودة عادة ما تعامل التسلسل والبنية كأوضاع مستقلة. عادةً ما تقوم طرق توليد البنية بتوليد ذرات السلسلة الرئيسية فقط. تتطلب الطرق التي تستهدف تصميم الذرات بالكامل عادةً استخدام نماذج خارجية للتبديل بين التنبؤ بالبنية وخطوات مقاومة الطي، وما إلى ذلك.
ولمعالجة هذه التحديات، اقترح فريق بحثي من جامعة كاليفورنيا في بيركلي، ومايكروسوفت للأبحاث، وجينينتك طريقة متعددة الوسائط لتوليد البروتين تسمى PLAID (انتشار البروتين الكامن المستحث)، والتي يمكنها تحقيق توليد متعدد الوسائط من خلال رسم الخرائط من وسائط بيانات أكثر ثراءً (مثل التسلسلات) إلى وسائط أكثر ندرة (مثل الهياكل البلورية).ولإثبات صحة هذا النهج، أجرى الباحثون تجارب على 2219 وظيفة من علم الجينات و3617 كائنًا حيًا عبر شجرة الحياة.على الرغم من عدم استخدام أي مدخلات هيكلية أثناء التدريب، إلا أن العينات المولدة تظهر جودة هيكلية قوية وتناسقًا.
البحث ذو الصلة يحمل عنوان "إنشاء بنية بروتينية كاملة الذرات من بيانات التدريب التسلسلي فقط" وتم تقديمه إلى المؤتمر الرائد ICLR 2025. كما قام "عراب الذكاء الاصطناعي" يانغ ليكون بإعادة نشر هذا الإنجاز على المنصة الاجتماعية.
عنوان مشروع PLAID مفتوح المصدر:
http://github.com/amyxlu/plaid
أبرز الأبحاث:
* مع التركيز على نموذج لغة البروتين الكبير ESMFold وتوليد بنية ذرية كاملة، اقترح الباحثون نموذج انتشار يمكن التحكم فيه ويمكنه توليد تسلسلات وبنى بروتينية ذرية كاملة في وقت واحد، ولا يتطلب سوى إدخال التسلسل أثناء التدريب.
* يستغل النهج المعلومات البنيوية المشفرة في الأوزان المدربة مسبقًا بدلاً من بيانات التدريب ويزيد من توفر تعليقات التسلسل لتوليد قابل للتحكم.
* على الرغم من استخدام نموذج ESMFold في البحث، إلا أنه يمكن تطبيق الطريقة على أي نموذج تنبؤ.
عنوان الورقة:
https://www.biorxiv.org/content/10.1101/2024.12.02.626353v1
يجمع المشروع المفتوح المصدر "awesome-ai4s" أكثر من مائة تفسير ورقي لـ AI4S ويوفر مجموعات وأدوات ضخمة من البيانات:
https://github.com/hyperai/awesome-ai4s
نظرة عامة سريعة على أبرز نتائج البحث
مجموعة البيانات
استخدم الباحثون إصدار سبتمبر 2023 من قاعدة بيانات Pfam، والتي تحتوي على 57،595،205 تسلسلًا و20،795 عائلة. يعتبر PLAID متوافقًا تمامًا مع قواعد بيانات التسلسل الأكبر مثل UniRef أو BFD (حوالي 2 مليار تسلسل)، ومع ذلك اختارت هذه الدراسة استخدام Pfam لأن مجال التسلسل الخاص به يحتوي على المزيد من العلامات البنيوية والوظيفية، مما يجعل تقييم المحاكاة الحاسوبية للعينات المولدة أكثر ملاءمة. بالإضافة إلى ذلك، احتفظ الباحثون بنحو 15% من البيانات للتحقق منها.
تتوفر أكواد UniRef للكائنات الحية التي تم اشتقاق مجالات Pfam منها من ملف Pfam-A.fasta المقدم على خادم Pfam FTP. قام الباحثون بتحليل جميع الكائنات الحية الفريدة في مجموعة البيانات، ووجدوا ما مجموعه 3617 كائنًا حيًا مختلفًا، ثم أجروا تجارب على هذه الكائنات الحية للتحقق من فعالية طريقة PLAID.
الهندسة المعمارية النموذجية
PLAID هو نموذج جديد لتوليد البروتينات متعدد الوسائط والذي يمكن التحكم فيه عن طريق الانتشار في الفضاء الكامن للنماذج التنبؤية.تظهر نظرة عامة على الطريقة في الشكل أدناه. باختصار، يتم تقسيمها إلى 4 خطوات:
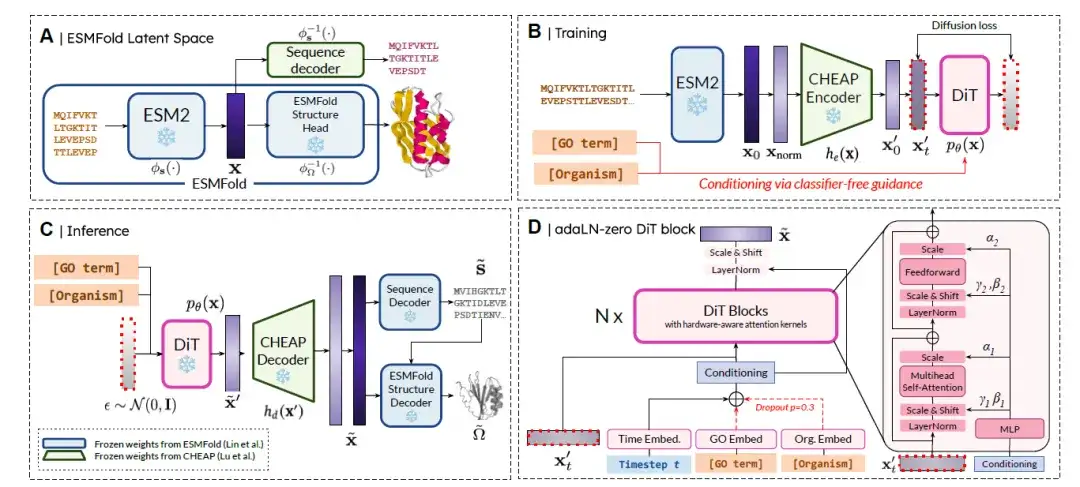
(أ) الفضاء الكامن لـ ESMFold:تمثل المساحة الكامنة p(x) التضمين المشترك للتسلسل والبنية.
(ب) تدريب الانتشار المحتمل:الهدف هو التعلم والعينة من pθ(x)، باتباع صيغة الانتشار. لتحسين كفاءة التعلم، يستخدم الباحثون مشفر CHEAP he(·) للحصول على التضمين المضغوط x′ = he(x)، بحيث يصبح هدف الانتشار هو أخذ العينات من pθ(he(x)).
(ج) الاستدلال:لالتقاط كل من التسلسل والبنية في وقت الاستدلال، نستخدم النموذج المدرب لعينة ˜x′ ∼ pθ(x′)، ثم نقوم بفك ضغطه باستخدام فك التشفير CHEAP للحصول على ˜x = hd(˜x′). يتم فك تشفير التضمين إلى تسلسل الأحماض الأمينية المقابل بواسطة فك تشفير التسلسل المجمد المدرب في CHEAP. يتم استخدام تسلسل هوية البقايا و˜x كمدخلات إلى فك تشفير البنية المجمدة المدرب في ESMFold للحصول على بنية الذرة بالكامل.
(د) هندسة كتلة DiT:استخدم الباحثون بنية المحول المنتشر (DiT) جنبًا إلى جنب مع كتلة DiT adaLN-zero لدمج المعلومات الشرطية. تم تضمين العلامات الوظيفية (أي مصطلحات GO) وعلامات فئة الكائن الحي دون استخدام إرشادات التصنيف.
نتائج الدراسة
أجرى الباحثون تحليلًا للجودة البنيوية والتنوع لأطوال البروتين المختلفة، وتظهر النتائج في الشكل أدناه.تحتوي العينات الأصلية للبروتين والعينات الناتجة عن PLAID على مقاييس متسقة بأطوال مختلفة.أظهر كل من ProteinGenerator وProtpardelle انهيارًا في الوضع عند أطوال معينة، بينما أظهر Multiflow انخفاضًا في التنوع في التسلسلات الأطول.
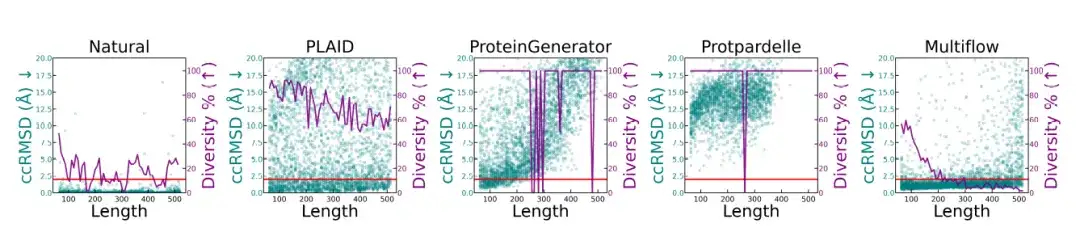
* يقارن هذا الشكل البروتينات الطبيعية وطرق التوليد المختلفة، مع إظهار الجودة البنيوية (ccRMSD، النقاط السماوية) والتنوع (الخط الأرجواني، الذي يتم قياسه كنسبة من المجموعات البنيوية الفريدة في العينة الإجمالية) للبروتينات بأطوال مختلفة (64-512 بقايا). الخط الأحمر يقع عند 2Å، مما يشير إلى عتبة التصميم)
بالإضافة إلى ذلك، بالمقارنة مع طريقة الأساس،إن التنوع الهيكلي الثانوي الذي يولده PLAID يشبه إلى حد كبير توزيع البروتينات الأصلية.كما هو موضح في الشكل أدناه: تظهر ProteinGenerator وProtpardelle وMultiflow انحرافات في توزيعات البنية الثانوية الخاصة بها، وعادةً ما تواجه نماذج توليد بنية البروتين الموجودة صعوبة في توليد عينات ذات محتوى عالٍ من صفائح β.
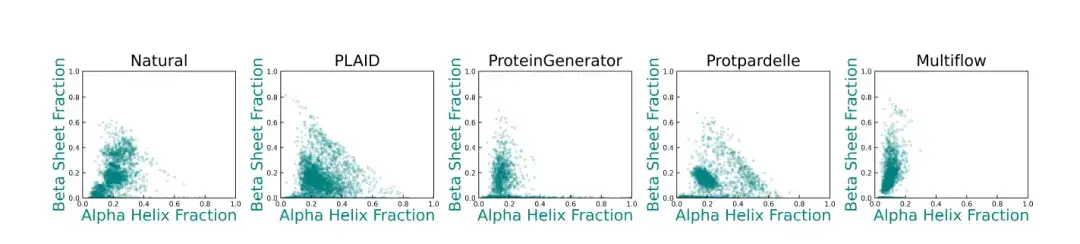
* يوضح هذا الشكل توزيع محتوى α-helix وβ-sheet للبروتينات الطبيعية والهياكل البروتينية الناتجة عن طرق مختلفة. تمثل كل نقطة بنية، وتمثل إحداثياتها نسبة بقايا اللولب ألفا (المحور السيني) ونسبة بقايا الصفيحة بيتا (المحور الصادي)
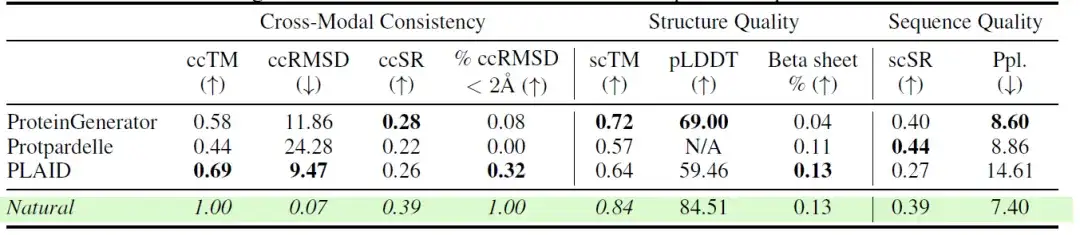
كما قام الباحثون بمقارنة أداء نماذج مختلفة عبر مقاييس متعددة للاتساق والجودة في مهمة توليد البروتين الذري بالكامل. وتظهر النتائج في الجدول التالي:تظهر العينات التي تم إنشاؤها بواسطة PLAID اتساقًا عاليًا بين التسلسل والبنية.
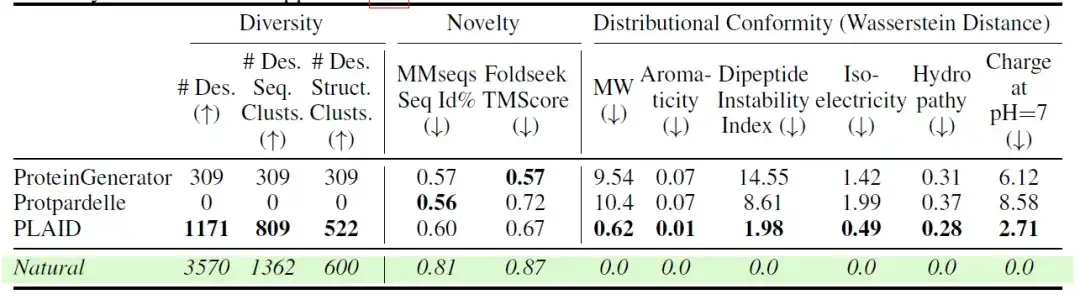
كما قام الباحثون أيضًا بتقييم التنوع والحداثة والطبيعية للنماذج المختلفة. وتظهر النتائج في الجدول التالي:من بين النماذج الذرية الكاملة، قام PLAID بإنشاء العينات الأكثر تفردًا وتصميمًا في كل من التسلسل والفضاء الهيكلي.
ومن الجدير التأكيد على أن PLAID يمكن توسيعه بسهولة ليشمل العديد من الوظائف اللاحقة ولا يقتصر على ESMFold، ولكن يمكن تطبيقه على أي نموذج تنبؤ.
الذكاء الاصطناعي يفتح آفاقًا جديدة لأبحاث البروتين
يتم استخدام المحول الانتشاري بشكل متزايد في المجال البيولوجي
يذكر هذا البحث أنه أثناء عملية بناء النموذج، استخدم الباحثون محول الانتشار (DiT) لأداء مهام إزالة الضوضاء.
المبدأ الأساسي لـ DiT هو تطبيق بنية المحول على نموذج الانتشار. عادةً ما تؤدي نماذج الانتشار إلى إفساد البيانات الأصلية عن طريق إضافة الضوضاء تدريجيًا ثم استعادة هذه البيانات من خلال التعلم النموذجي. يعمل DiT على تعزيز القدرة التوليدية للنموذج من خلال إدخال كتل المحول (مثل تطبيع الطبقة التكيفية، والانتباه المتبادل، وما إلى ذلك) في نموذج الانتشار.
لقد حققت DiT في السنوات الأخيرة تقدماً كبيراً في مجال إنتاج الصور والفيديو. إن البنية الأساسية للنماذج المتطورة مثل Sora هي DiT.في مجال الطب الحيوي، أصبح تطبيق المحول الانتشاري أكثر وأكثر اتساعًا. ويمكن أن يساعد الباحثين على فحص جزيئات الأدوية المحتملة بسرعة والتنبؤ بنشاطها البيولوجي. ويمكنه أيضًا المساعدة في المهام المعقدة مثل تحليل تسلسل الجينات والتنبؤ ببنية البروتين، مما يوفر أداة قوية لأبحاث علوم الحياة.باستخدام إزالة الضوضاء من البروتين كمثال، يمكن لتقنية DiT التقاط العلاقات المعقدة بين التسلسل والبنية. وهذا يعني أنه من خلال آلية الاهتمام الذاتي العالمية للمحول، فإنه يمكنه نمذجة العلاقة التفاعلية المعقدة بين تسلسل البروتين وبنيته بشكل فعال، ثم استخدام العملية العكسية لنموذج الانتشار للتنبؤ بالناقل الكامن الخالي من الضوضاء في كل خطوة زمنية، واستعادة بنية وتسلسل البروتين تدريجيًا من الضوضاء.
بالنسبة لهذه الورقة البحثية على وجه التحديد، توفر DiT خيارات أكثر مرونة للضبط الدقيق للتعامل مع الوسائط المدخلة المختلطة، خاصة وأن نماذج التنبؤ ببنية البروتين بدأت في دمج الأحماض النووية ومجمعات الربيطة الجزيئية الصغيرة. علاوة على ذلك، يتيح هذا النهج استخدام البنية التحتية لتدريب المحولات بشكل أفضل.
وفي التجارب المبكرة، وجد الباحثون أيضًا أن تخصيص الذاكرة المتاحة لنماذج DiT الأكبر حجمًا كان أكثر كفاءة من استخدام الاهتمام الذاتي الثلاثي. ويستخدم نموذج تدريب خوارزمية التحسين الذي تم تنفيذه بواسطة xFormers، وقد حقق تحسنًا في السرعة بمقدار 55.8% وانخفاضًا بمقدار 15.6% في استخدام ذاكرة وحدة معالجة الرسومات في اختبار معيار مرحلة الاستدلال.
التعلم الآلي يجعل البروتينات المخصصة "حلمًا يتحقق"
ويمكن القول إن البحث المذكور أعلاه من جامعة كاليفورنيا في بيركلي يشكل خطوة مهمة أخرى إلى الأمام في تخصيص البروتين. نحن نعلم أن البروتينات تتكون عادة من 20 حمضًا أمينيًا مختلفًا، والتي يمكن اعتبارها اللبنات الأساسية للحياة.وبسبب بنيتها المعقدة للغاية، كان من غير الممكن للعلماء قبل عقود من الزمن التنبؤ بالبنية الثلاثية الأبعاد للبروتينات وتصميم بروتينات جديدة للاستخدام البشري. ومع ذلك، فإن التقدم السريع في مجال التعلم الآلي في السنوات الأخيرة جعل حلم تصميم البروتينات المخصصة ممكنًا تدريجيًا.
بالإضافة إلى AlphaFold الشهير، هناك بعض التقدم البحثي الذي يستحق الاهتمام أيضًا:
في نوفمبر 2024، نجح فريق من مختبر أرجون الوطني التابع لوزارة الطاقة الأمريكية في تطوير إطار عمل حوسبة مبتكر يسمى MProt-DPO.يجمع الإطار بين تكنولوجيا الذكاء الاصطناعي وأفضل أجهزة الكمبيوتر العملاقة في العالم، مما يمثل عصرًا جديدًا في تصميم البروتين. وباستخدام MProt-DPO كمثال، صمم العلماء نوعًا جديدًا من الإنزيمات يمكنه تحفيز التفاعلات الكيميائية بكفاءة في ظل ظروف محددة. وبالمقارنة مع طرق التصميم السابقة، تم تحسين كفاءة تفاعل الإنزيم الجديد بنحو 30%، وهو ما لا يؤدي إلى تسريع التقدم التجريبي فحسب، بل يوفر أيضًا المزيد من الاحتمالات للتطبيقات الصناعية. علاوة على ذلك، فإن التطبيق الناجح لـ MProt-DPO يفتح أيضًا أفكارًا جديدة لتصميم البروتينات المضادة للفيروسات. تم نشر نتائج البحث ذات الصلة في مجلة IEEE Computer Society تحت عنوان "MProt-DPO: كسر حاجز ExaFLOPS لسير عمل تصميم البروتين متعدد الوسائط مع تحسين التفضيل المباشر".
عنوان الورقة:
https://www.computer.org/csdl/proceedings-article/sc/2024/529100a074/21HUV88n1F6
الجيوب البروتينية هي مواقع على البروتينات مناسبة للارتباط بجزيئات محددة. يعد تصميم جيب البروتين أحد الأساليب المهمة في عملية تخصيص البروتينات. في ديسمبر 2024، صممت جامعة العلوم والتكنولوجيا في الصين وزملاؤها خوارزمية التوليد العميق PocketGen.يمكن إنشاء تسلسلات وهياكل جيب البروتين على أساس إطار البروتين والجزيئات الصغيرة المرتبطة. وتظهر التجارب أن المؤشرات مثل تقارب نموذج PocketGen والعقلانية البنيوية تتجاوز الطرق التقليدية، كما تم تحسين الكفاءة الحسابية بشكل كبير. وقد نُشرت نتائج البحث ذات الصلة في مجلة Nature Machine Intelligence تحت عنوان "التوليد الفعال للجيوب البروتينية باستخدام PocketGen".
عنوان الورقة:
https://www.nature.com/articles/s42256-024-00920-9

في المستقبل، ومع المزيد من تطبيق الذكاء الاصطناعي في مجال البروتين، أعتقد أن الناس سيكون لديهم فهم أعمق لأسرار البنية المكانية للبروتين.
مراجع:
1.https://www.biorxiv.org/content/10.1101/2024.12.02.626353v1
2.https://mp.weixin.qq.com/s/_5_L7bvl-vHtls8gBbfSmQ
3.https://mp.weixin.qq.com/s/sfrm2rj_8kH0JA2vu4NmTw
4.http://www.news.cn/globe/20241014/f7137840e56340f081f9eb819d87ba40/c.html
5.http://www.bfse.cas.cn/yjjz/202412/t20241212_5042432.html
6.https://www.sohu.com/a/826241274_12








