Command Palette
Search for a command to run...
تم الاختيار في ICML! فريق معهد ماساتشوستس للتكنولوجيا يحقق اختراقًا جديدًا يعتمد على AlphaFold، ويكشف عن التنوع الديناميكي للبروتينات
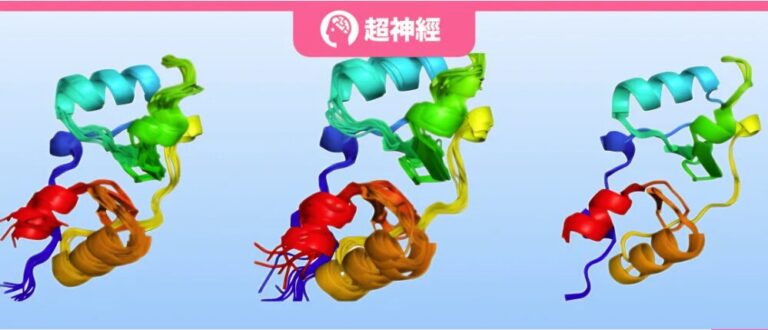
باعتبارها مكونًا مهمًا للكائنات الحية، فإن البروتينات لها حالات مختلفة وتعتمد هياكل ثلاثية الأبعاد معقدة تعتمد على مجموعات هيكلية مختلفة من الحركة الجماعية أو التقلبات غير المنظمة لأداء وظائف بيولوجية غنية. على سبيل المثال، تعتبر التغيرات التكوينية للبروتينات ذات أهمية بالغة لوظائف الناقلات والقنوات والإنزيمات، في حين تساعد خصائص التركيبة المتوازنة في التحكم في قوة وانتقائية التفاعلات الجزيئية.
في السنوات الأخيرة، حققت أساليب التعلم العميق مثل AlphaFold نجاحًا كبيرًا في نمذجة الحالة الفردية للبروتينات، لكنها لا تستطيع تفسير التباين التكويني. لذا، بالنسبة لعلماء الأحياء البنيوية،كيف نضمن التنبؤ الدقيق لهيكل واحد مع الكشف عن التركيبات الهيكلية المحتملة؟إنها مشكلة صعبة وتحتاج إلى حل عاجل.
مؤخرًا، قام فريق بحثي من معهد ماساتشوستس للتكنولوجيا بدمج طرق أخذ العينات الجديدة من AlphaFold وESMFold وقدم منظورًا جديدًا لمراقبة وفهم المساحة التكوينية للبروتينات من خلال تقنية مطابقة التدفق.
توضح هذه الدراسة أداء متغيرات مطابقة التدفق AlphaFlow وESMFlow في سيناريوهين مختلفين.تم في النهاية ضبط النموذج على قاعدة بيانات البروتين وتدريبه بشكل أكبر على مجموعة بيانات ATLAS، وكلاهما أظهر أداءً متفوقًا، ليس فقط تجاوز خط الأساس التقليدي لـ MSA في التنبؤ بالمرونة التكوينية ونمذجة توزيع الموضع الذري، ولكن أيضًا تحقيق تقدم كبير في تكرار ملاحظات المجموعة ذات الترتيب الأعلى.
تم اختيار البحث ذي الصلة، بعنوان "AlphaFold Meets Flow Matching for Generating Protein Ensembles"، لمؤتمر ICML 2024، وهو المؤتمر الأكاديمي الأهم في مجال الذكاء الاصطناعي.

عنوان الورقة:
https://openreview.net/forum?id=rs8Sh2UASt
يجمع المشروع المفتوح المصدر "awesome-ai4s" أكثر من مائة تفسير ورقي لـ AI4S ويوفر مجموعات وأدوات ضخمة من البيانات:
https://github.com/hyperai/awesome-ai4s
مجموعة البيانات: استنادًا إلى مجموعات بيانات PDB وATLAS لضمان نزاهة النتائج التجريبية
كما نعلم جميعًا، تم تطوير AlphaFold وتدريبه بطريقة شاملة استنادًا إلى الهياكل الموجودة في قاعدة بيانات البروتين، بينما استخدم ESMFold التضمينات من نموذج لغة البروتين (PLM) كمدخلات. لذلك،استخدمت هذه الدراسة بشكل أساسي مجموعة بيانات PDB ومجموعة بيانات MD.
أولاً، لبناء مجموعة اختبار من البروتينات غير المتجانسة هيكليًا من قاعدة بيانات البروتينات، استخدمنا قاعدة بيانات توضيح SIFTS ورسم الخرائط على مستوى البقايا من سلاسل قاعدة بيانات البروتينات إلى تسلسلات UniProt المرجعية، وربط كل سلسلة متبقية بشظية. وبعد ذلك، تم ربط جميع أجزاء المجموعات بشكل كامل استنادًا إلى عتبة تشابه جاكارد البالغة 0.75، مع التعامل مع كل مجموعة ناتجة باعتبارها بروتينًا فريدًا.وقد أدى ذلك إلى إنتاج 75000 بروتين.
بالإضافة إلى ذلك، جمعت الدراسة:
* البروتينات التي لم ترسل السلاسل قبل الموعد النهائي لتدريب AlphaFold، ولكنها أودعت 2-30 سلسلة بعد الموعد النهائي؛
* البروتينات ذات أطوال تتراوح بين 256 إلى 768 بقايا؛
* البروتينات التي تحتوي على مجموعتين هيكليتين على الأقل عندما كانت عتبة التكتل المتسلسل 0.85 lDDT-Cα متماثلة واتصال كامل.
وأخيرا، تم الحصول على 563 بروتينًا ممثلة بـ 2843 سلسلة.قام الباحثون باستخراج 100 بروتين ممثلة بـ 500 سلسلة لتشكيل مجموعة اختبار.
ثانيًا، قام الباحثون ببناء مجموعة بيانات ATLAS استنادًا إلى مجموعة بيانات MD.يتكون الأخير من 1390 بروتينًا تم اختيارها بناءً على تصنيف مجال ECOD.بالنسبة لكل بروتين، توفر مجموعة البيانات ثلاث عمليات محاكاة متكررة بطول 100 نانوثانية، وتحتوي كل محاكاة على 10000 إطار. لتدريب هذه المسارات والتحقق من صحتها، قمنا أولاً بإنشاء MSAs لجميع إدخالات ATLAS البالغ عددها 1390 باستخدام التسلسلات المقدمة وخط أنابيب ColabFold MMSeqs2.
وبعد ذلك، اختار الباحثون بشكل عشوائي 300 تكوين من خط أنابيب التدريب، باستخدام 1 مايو 2018 و1 مايو 2019 كمواعيد نهائية للتدريب والتحقق على التوالي، وحصلوا في النهاية على 1265/39/82 مجموعة من مجموعات التدريب والتحقق والاختبار.

بناء النموذج: استخدام AlphaFold كنموذج لإزالة الضوضاء لإجراء مطابقة التدفق على مجموعات البروتين
نظرًا للتحديات الكبيرة المتمثلة في إعادة تطوير نموذج توزيع بنفس دقة وقدرات التعميم مثل AlphaFold، تستفيد هذه الدراسة من التطورات المفاهيمية الحديثة في النماذج التوليدية.من السهل تقريبًا إعادة استخدام AlphaFold كنموذج توليدي.

حتى الآن، تقوم هياكل نموذج الانتشار النموذجية من النص إلى الصور تقريبًا جميعها بنمذجة التوزيع الشرطي p(x | s) للصورة x المشروطة بإشارة نصية s. في قلب هذه النماذج توجد شبكة عصبية لإزالة الضوضاء تستقبل صورة بها ضوضاء ومطالبة نصية وتتنبأ بصورة نظيفة.
وبناءً على هذه الظروف، يتم تدريب هذه النماذج عادةً باستخدام هدف الخطأ التربيعي المتوسط البسيط (MSE). على نحو مماثل، يمكن تحويل متنبئ بنية البروتين المدرب باستخدام دالة خسارة تشبه الانحدار مثل AlphaFold أو ESMFold إلى نموذج خالٍ من الضوضاء ببساطة عن طريق توفير مدخلات بنية ضوضائية إضافية. بفضل هذه التعديلات المعمارية، يمكن لهذه الدراسة إدراج AlphaFold وESMFold بشكل أكبر في أي إطار عمل للنمذجة التوليدية القائمة على إزالة الضوضاء التكرارية.
تعتقد هذه الدراسة أن تصميم إطار عمل توليد مطابقة التدفق يعادل اختيار مسار الاحتمال الشرطي pt(x | x1) وحقل المتجه المقابل له ut(x | x1). لذلك، تحدد هذه الدراسة شبكة عصبية معاد معاملتها x1(x, t; θ) عن طريق أخذ عينات من الضوضاء x0 من q(x0) ومعالجتها خطيًا بنقطة البيانات x1 لتحديد مسار الاحتمالية الشرطية.وبالتالي، يتم استخدام بنية AlphaFold كنموذج لإزالة الضوضاء.
لتطبيق مطابقة التدفق على هياكل البروتين، تصف الدراسة أيضًا الهيكل من خلال إحداثيات ثلاثية الأبعاد لكربونات بيتا (كربون ألفا للجليسين): x ∈ R^N×3. ويضمن هذا أيضًا أن يكون الإدخال إلى الشبكة العصبية دائمًا عبارة عن بنية ثلاثية الأبعاد تشبه البوليمر، وقابلة للتصديق من الناحية الفيزيائية.
نظرًا لأن إطار مطابقة التدفق يتضمن تحديد عمليات الضوضاء وعكسها، فإنه يحتوي على العديد من أوجه التشابه مع الانتشار التوافقي لهياكل البروتين، حيث يتقارب كلاهما إلى نفس التوزيع المسبق. ومع ذلك، كإطار أكثر عمومية،توفر مطابقة التدفق ميزتين رئيسيتين:
أولاً،يتقارب الانتشار التوافقي مع التوزيع السابق فقط في حد زمني لا نهائي، ويعتمد معدل التقارب على أبعاد البيانات، أي حجم البروتين. ويؤدي هذا إلى تحول في التوزيع في وقت الاستدلال عند التدريب فقط على المحاصيل ذات الحجم الصغير نسبيًا.
ثانيًا،توفر مطابقة التدفق طريقة سهلة للتعامل مع البقايا المفقودة الشائعة جدًا في قاعدة بيانات البروتين عن طريق حذفها ببساطة. على النقيض من ذلك، فإن الانتشار التوافقي يخلق اعتماديات بين المواضع الذرية، مما يستلزم استيفاء البيانات للبقايا المفقودة.

أخيرًا، قامت الدراسة بضبط جميع أوزان AlphaFold وESMFold على PDB استنادًا إلى إطار عمل مطابقة العملية، وكانت مواعيد التدريب النهائية لـ AlphaFold وESMFold المستخدمة هي 1 مايو 2018 و1 مايو 2020 على التوالي. وفي نهاية هذه المرحلة من التدريب، حصلت الدراسة على متغيرات مطابقة التدفق لـ AlphaFold وESMFold،وأطلقوا عليه اسم AlphaFLOW وESMFLOW.
لتقييم القدرة على التعلم من مجموعات الديناميكية الجزيئية، تم ضبط النموذجين بشكل أكبر على مجموعة بيانات ATLAS التي تحتوي على عمليات محاكاة الديناميكية الجزيئية لجميع الذرات. بعد التدريب على 43000 و27000 مثال إضافي على التوالي،حصلت الدراسة على متغيرات نموذجية خاصة بـ MD - AlphaFLOW-MD و ESMFLOW-MD.
النتائج التجريبية: الأداء يفوق أداء الطرق التقليدية وله آفاق تطبيق واسعة في مجال البيولوجيا البنيوية
قام الباحثون أولاً بتقييم قدرات AlphaFLOW وESMFLOW للتكوينات المتنوعة للبروتينات المودعة في PDB.
ولتحقيق هذه الغاية، قامت الدراسة ببناء مجموعة اختبار تحتوي على 100 بروتين مع وجود أدلة على وجود سلاسل متعددة وتباين تكويني تم إيداعها بعد الموعد النهائي لتدريب AlphaFold (1 مايو 2018)، وتم تقييمها لثلاثة مؤشرات رئيسية: الدقة، والتذكير، والتنوع.

تظهر النتائج أن AlphaFLOW يشبه أخذ العينات الفرعية من MSA في أن كليهما يزيد من تنوع التوقعات على حساب الدقة، ولكن بالمقارنة مع أخذ العينات الفرعية من MSA، فإن متغيرات AlphaFLOW تتبع جبهات باريتو بشكل أفضل بكثير.
من حيث الدقة والتذكير،يُظهر AlphaFLOW سلوكًا مشابهًا جدًا لعينات فرعية من MSA.ومن المثير للدهشة إلى حد ما أن أيًا من النهجين لا يعمل على تحسين التذكر الشامل بشكل كبير مقارنةً بـ AlphaFold الأساسي.
بشكل عام، تعتبر دقة ESMFold وESMFLOW أقل نسبيًا مقارنة بعائلة أساليب AlphaFold. ومع ذلك، فإن ESMFLOW قادر على حقن قدر كبير من التنوع نسبة إلى ESMFold الأساسي.وتحسين التذكر مع عدم التضحية تقريبًا بالدقة.
بالإضافة إلى ذلك، أظهر تحليل RMWD لهذه الدراسة أن AlphaFlow كان أفضل قليلاً من AlphaFold في التنبؤ بالموضع المتوسط للذرات وأفضل بشكل كبير من أخذ العينات الفرعية MSA من حيث تباين النمذجة.

قامت هذه الدراسة بتقييم قدرة AlphaFLOW وESMFLOW على توليد مجموعات MD بالوكالة لمجموعة اختبار مكونة من 82 بروتينًا في قاعدة بيانات ATLAS. استخدمت هذه الدراسة عينات منفصلة من كل طريقة وفحصت مدى تشابه العينة مع مجموعة مرضى التصلب المتعدد باستخدام سلسلة من التقييمات.
وتظهر النتائج أنيحقق AlphaFLOW-MD تحسينات كبيرة في التشابه، متجاوزًا أداء أخذ العينات الفرعية MSA.

نظرًا لأن MD يعتبر القيمة الحقيقية، فإن تشغيله حتى التقارب يعد مكلفًا. لذلك، تقوم هذه الدراسة بتحليل ما إذا كان AlphaFLOW قادرًا على تقديم نتائج أفضل في ظل ميزانية حسابية محدودة مكافئة، على سبيل المثال، في ساعات وحدة معالجة الرسومات. ولتحقيق هذه الغاية، قامت الدراسة بتقليص عدد العينات المستخرجة من AlphaFLOW (من 250 إلى 4) وتقصير طول مسار MD (من 100ns إلى 160ps).
وتظهر النتائج أن جودة مجموعة AlphaFLOW تظل ثابتة، ولكن مسارات MD تستغرق وقتًا أطول للوصول إلى نفس مستوى الجودة أو تجاوزه.
تبرز ثلاثة نماذج رئيسية للتدريب المسبق للبروتين العام، ومجال علم الأحياء البنيوي مليء بالحيوية
في السنوات القليلة الماضية، كانت البروتينات والذكاء الاصطناعي تتصادم باستمرار لخلق شرارات جديدة.في الوقت الحاضر، شكل التدريب المسبق الشامل للبروتينات وضعًا جديدًا يتكون من ثلاثة ركائز.وهذا يشمل سلسلة DeepMind Alphafold، وسلسلة David Baker's RoseTTAFold، وسلسلة Meta ESM. وبناء على هذه النماذج الثلاثة، بدأت نتائج البحوث العلمية ذات الصلة تتفجر. وفي النصف الأول من عام 2024 وحده، تم نشر العديد من نتائج الأبحاث في مجلات مرموقة مثل Nature وScience.
في مارس 2024، نشر باحثون من كلية الطب بجامعة نورث كارولينا، وجامعة كاليفورنيا في سان فرانسيسكو، وجامعة ستانفورد، وجامعة هارفارد دراسة في مجلة ساينس تؤكد أنيمكن أن تساعد هياكل AlphaFold2 المتوقعة في توجيه اكتشاف الأدوية في المستقبل.ووجد فريق البحث أن AlphaFold2 أظهر فائدة عملية كبيرة في علم الأحياء البنيوي، وتصميم البروتين، والتفاعلات، والتنبؤ بالهدف، والتنبؤ بالوظيفة، والآليات البيولوجية، وكان قادرًا على البحث عن أدوية جديدة محتملة من خلال فحص مليارات المركبات ومطابقة المكتبات مع هياكل البروتين.
في مايو 2024، أصدر فريق Google DeepMind برنامج AlphaFold 3 في مجلة Nature، مما أدى إلى توسيع نطاق التكنولوجيا إلى ما هو أبعد من طي البروتينات والتنبؤ بدقة ببنية وتفاعلات جزيئات الحياة مثل البروتينات والحمض النووي والحمض النووي الريبوزي والربيطة بدقة غير مسبوقة. هذا يعنى،سيعمل AlphaFold 3 على تسريع تصميم الأدوية والأبحاث الجينومية بشكل أكبر.تدشين عصر جديد من الذكاء الاصطناعي في علم الأحياء الخلوي.
مع إصدار AlphaFold 3،تمكنت سلسلة Alphafold أخيرًا من بناء أساس ذري بالكامل.وعلى نحو مماثل، أطلقت سلسلة RoseTTAFold بنجاح أيضًا RoseTTAFold All-Atom في النصف الأول من هذا العام، محققة القدرة على تقديم تنبؤات معقولة بشأن التعديلات التساهمية للبروتين وتجميع سلاسل متعددة من الأحماض النووية والجزيئات الصغيرة.
بمساعدة Alphafold3 وRoseTTAFold All-Atom، يتمكن الباحثون من الاستفادة الكاملة من قوة الخيال. على سبيل المثال، في يونيو 2024، نشر فريق بحثي دولي ورقة بحثية في مجلة Nature Biotechnology، توضح كيفية استخدام استراتيجية الجمع بين AlphaFold 3 وRoseTTAFold All-Atom لتصميم نوع جديد من هيكل البروتين بنجاح والذي يمكنه توصيل الأدوية بشكل أكثر فعالية مباشرة إلى الخلايا المريضة، وبالتالي تحسين التأثيرات العلاجية وتقليل الآثار الجانبية. ويمثل هذا الاكتشاف خطوة قوية إلى الأمام في تطبيق الذكاء الاصطناعي في الطب الدقيق.
لسوء الحظ، في أغسطس 2023، قامت Meta بحل فريق ESMFold ووجهت اهتمامها الكامل إلى الترويج لتسويق الذكاء الاصطناعي. ولكن الأبحاث حول سلسلة ESM لم تتوقف. على سبيل المثال، حقق النموذج تقدمًا مهمًا في مجال نمذجة لغة البروتين وقدم حلًا موحدًا للنمذجة يدمج المعلومات متعددة المقاييس. ومن الجدير بالذكر أن هذا هو أول نموذج لغوي بروتيني مدرب مسبقًا يمكنه معالجة معلومات الأحماض الأمينية والمعلومات الذرية.
ومن هذا يمكن أن نرى أنفي العصر الجديد حيث أصبحت سلسلة Alphafold وسلسلة RoseTTAFold وسلسلة ESM على قدم المساواة مع بعضها البعض،وسوف يصبح الجمع بين الذكاء الاصطناعي وأبحاث البروتين أقرب، الأمر الذي لن يؤدي فقط إلى تسريع فهمنا لبنية البروتين ووظيفته، بل سيجلب أيضًا تغييرات ثورية في علاج الأمراض وتطوير الأدوية وتطبيقات التكنولوجيا الحيوية. مع التطور السريع الذي أحدثته تكنولوجيا الذكاء الاصطناعي، أصبح مجال علم الأحياء البنيوي أكثر حيوية، كما يتكشف فصل جديد في مجال الطب الحيوي ببطء.








