Command Palette
Search for a command to run...
نموذج الخلية المكون من 100 مليون معلمة موجود هنا! نشر فريق جامعة تسينغهوا في مجلة Nature بحثًا بعنوان "النمذجة المتزامنة لـ 20 ألف جين"
في السنوات الأخيرة، أصبحت النماذج المدربة مسبقًا على نطاق واسع رائدة في موجة جديدة من الذكاء الاصطناعي. يستخرج "النموذج الكبير" قواعد عميقة المستوى من بيانات واسعة النطاق ومتعددة المصادر، ويمكن بعد ذلك أن يعمل بمثابة "نموذج أساسي" لخدمة مجموعة متنوعة من المهام في مجالات مختلفة. على سبيل المثال، أتقنت نماذج اللغة الكبيرة القدرة على فهم اللغة والتعرف عليها من خلال التعلم من كميات كبيرة من بيانات النصوص، مما أحدث ثورة في النموذج في مجال معالجة اللغة الطبيعية.
وعلى نحو مماثل، في مجال علوم الحياة، تمتلك الكائنات الحية أيضًا "لغتها الأساسية" الخاصة بها - فالخلايا هي الوحدات البنيوية والوظيفية الأساسية لجسم الإنسان.إذا تمت مقارنة قيم التعبير الجيني والحمض النووي الريبوزي والبروتين مع "الكلمات"، يتم دمجها معًا لتشكيل الجملة "خلية".وبالتالي، إذا تمكنا من تطوير نموذج خلية الذكاء الاصطناعي على أساس "لغة" الخلية، فمن المأمول أن يوفر ذلك نموذجًا بحثيًا جديدًا وأدوات بحثية ثورية لعلوم الحياة والطب.
لكن،هناك حاليًا ثلاثة تحديات رئيسية في تدريب بيانات الخلية الواحدة واسعة النطاق:
* يجب أن تغطي بيانات التدريب المسبق للتعبير الجيني المناظر الطبيعية للخلايا ذات الحالات والأنواع المختلفة. في الوقت الحالي، يتم تنظيم معظم بيانات تسلسل الحمض النووي الريبي للخلية الواحدة (scRNA-seq) بشكل فضفاض، ولا تزال قاعدة البيانات الشاملة والكاملة مفقودة؛
* أثناء التدريب، يواجه المحولون التقليديون صعوبة في معالجة "الجمل" المكونة من ما يقرب من 20 ألف جين مشفر للبروتين؛
* تختلف بيانات scRNA-seq من تقنيات ومختبرات مختلفة في عمق التسلسل، مما يمنع النموذج من تعلم تمثيلات الخلايا والجينات الموحدة والمعبرة.
ولمعالجة هذه التحديات،تعاون في البحث البروفيسور تشانغ شيويه غونغ، مدير مختبر النموذج الأساسي للحياة في قسم الأتمتة بجامعة تسينغهوا، والبروفيسور ما جيان تشو من قسم الإلكترونيات/AIR، والدكتور سونغ لي من التكنولوجيا الحيوية.في يونيو 2024، تم نشر ورقة بحثية بعنوان "نموذج أساسي واسع النطاق لنسخ الجينات أحادية الخلية" في مجلة Nature Methods.
تقدم الورقة البحثية نموذجًا خلويًا كبيرًا يسمى scFoundation، والذي يمكنه معالجة حوالي 20 ألف جين في وقت واحد.وباعتباره نموذجًا أساسيًا، فإنه يوضح تحسينات أداء متميزة في مجموعة متنوعة من المهام الطبية الحيوية اللاحقة مثل تعزيز عمق تسلسل الخلايا، والتنبؤ باستجابة الخلايا للأدوية، والتنبؤ باضطراب الخلايا، مما يوفر نموذجًا جديدًا للذكاء الاصطناعي في أبحاث الخلية الواحدة.
أبرز الأبحاث:
تم تدريب نموذج خلية scFoundation على أساس بيانات التعبير الجيني من 50 مليون خلية، ويحتوي على 100 مليون معلمة، ويمكنه معالجة ما يقرب من 20000 جين في وقت واحد* يستخدم النموذج تصميمًا غير متماثل لتقليل التحديات الحسابية والذاكرة* يوفر النموذج أفكارًا بحثية جديدة لاستنتاج شبكة الجينات وتحديد عوامل النسخ

عنوان الورقة:
https://www.nature.com/articles/s41592-024-02305-7
يجمع مشروع المصدر المفتوح "awesome-ai4s" أكثر من 100 تفسير لورقة AI4S ويوفر أيضًا مجموعات بيانات وأدوات ضخمة:
https://github.com/hyperai/awesome-ai4s
مجموعات البيانات: بناء مجموعة بيانات شاملة من خلية واحدة
قام الباحثون ببناء مجموعة بيانات شاملة للخلية الواحدة من خلال جمع كل بيانات موارد الخلية الواحدة المتاحة للجمهور.وتشمل هذه الأدوات مجموعة التعبير الجيني (GEO)، وبوابة الخلية الواحدة، وHCA، ومشروع الجينوم البشري (hECA)، وبيانات أوميكس الخلية الواحدة البشرية المتكاملة بعمق (DISCO)، وقاعدة بيانات مختبر الأحياء الجزيئية الأوروبي - معهد المعلوماتية الحيوية الأوروبي (EMBL-EBI)، وما إلى ذلك.
* عنوان تنزيل GEO:https://www.ncbi.nlm.nih.gov/geo/
* رابط تحميل بوابة الخلية الواحدة:https://singlecell.broadinstitute.org/single_cell
* عنوان تنزيل HCA:https://data.humancellatlas.org/
* عنوان تنزيل EMBL-EBI:https://www.ebi.ac.uk/
قام الباحثون بربط كافة البيانات بقائمة جينية تضم 19264 جينًا مشفرًا للبروتين وجينًا مشتركًا للميتوكوندريا حددتها لجنة تسمية جينات HUGO. بعد مراقبة جودة البيانات،تم الحصول على أكثر من 50 مليون بيانات من تسلسل scRNA البشري للتدريب المسبق.
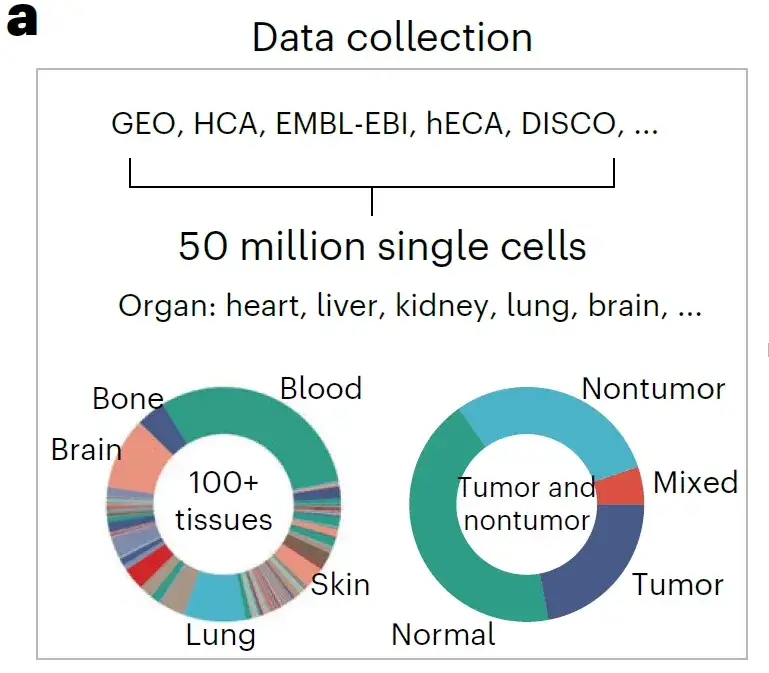
إن مصادر البيانات الوفيرة تقوم ببناء مجموعات بيانات مسبقة التدريب غنية بالأنماط البيولوجية. من الناحية التشريحية، يشمل أكثر من 100 نوع من الأنسجة، ويغطي مجموعة واسعة من الأمراض والأورام والحالات الطبيعية، وكما هو موضح في الشكل أعلاه، يشمل جميع أنواع الخلايا البشرية وحالاتها المعروفة تقريبًا.
هندسة النموذج: بناء نموذج scFoundation يحتوي على 100 مليون معلمة
يحتوي نموذج scFoundation الذي طوره الباحثون على ما يقرب من 100 مليون معلمة، ويعتبر مقياس المعلمات والتغطية الجينية ومقياس البيانات "من بين الأفضل" في مجال الخلية الواحدة.
تصميم النموذج
قام الباحثون بتطوير نموذج xTrimoGene باعتباره النموذج الأساسي لـ scFoundation، وهو نموذج قابل للتطوير يعتمد على المحول ويتضمن وحدة تضمين وهيكل ترميز وفك ترميز غير متماثل.
من بينها، تقوم وحدة المتجه بتحويل قيم التعبير الجيني المستمر إلى متجهات عالية الأبعاد قابلة للتعلم، مما يضمن الحفاظ على قيم التعبير الأصلية بالكامل؛ يأخذ المشفر الجينات التعبيرية غير الصفرية وغير المقنعة كمدخلات، ويستخدم كتلة محول الفانيليا ويحتوي على عدد كبير من المعلمات؛ يقوم فك التشفير بأخذ جميع الجينات كمدخلات، ويستخدم كتلة أداء، ويحتوي على عدد صغير نسبيًا من المعلمات.
يقلل هذا التصميم غير المتماثل من التحديات الحسابية والذاكرة مقارنة بالهندسة المعمارية الأخرى.تظهر البيانات أن الوحدة تتطلب فقط 3.4% من نموذج اللغة التقليدية Transformer مع الحفاظ على نفس مقياس المعلمات.
مهام ما قبل التدريب
قام الباحثون بتصميم مهمة تدريب مسبقة تسمى نمذجة RDA (قراءة الوعي بالعمق).هذا امتداد لنموذج اللغة المقنعة الذي يأخذ في الاعتبار التباين العالي في عمق التسلسل في البيانات واسعة النطاق.
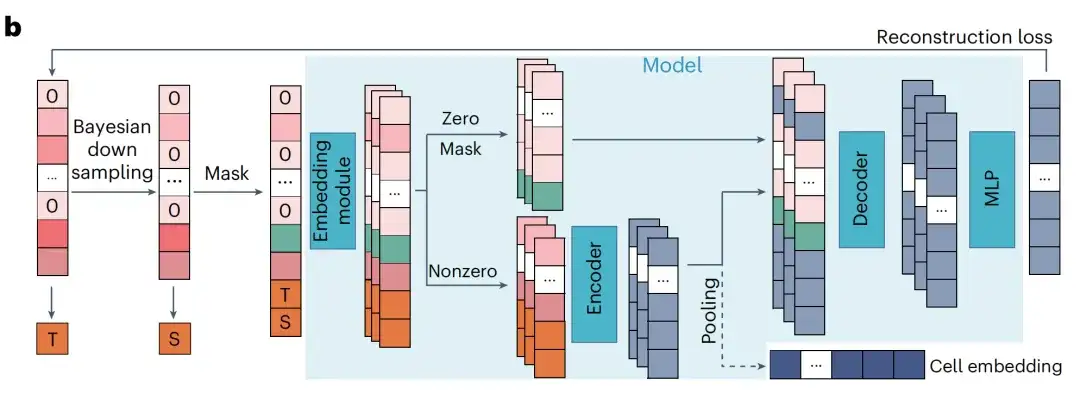
في نمذجة RDA، يتنبأ النموذج بتعبير الجينات المقنعة استنادًا إلى جينات سياق الخلية. اعتبر الباحثون العدد الإجمالي بمثابة عمق التسلسل للخلية وحددوا مقياسين للعدد الإجمالي: T (الهدف) وS (المصدر)، والتي تمثل العدد الإجمالي للعينة الأصلية وعينة الإدخال، على التوالي. قام الباحثون بإخفاء الجينات المعبر عنها وغير المعبر عنها بشكل عشوائي في عينات الإدخال وسجلوا مؤشراتها.
ثم يستخدم النموذج عينة الإدخال المقنعة والمقياسين للتنبؤ بقيمة التعبير للعينة الأصلية عند الفهرس المقنع. يتيح هذا للنموذج المدرب مسبقًا ليس فقط التقاط العلاقات الجينية داخل الخلايا، بل أيضًا تنسيق الخلايا ذات أعماق التسلسل المختلفة. أثناء الاستدلال، يقوم الباحثون بإدخال التعبير الجيني الخام للخلايا في النموذج المدرب مسبقًا وتعيين T أعلى من إجمالي عددهم S لتوليد قيم التعبير الجيني بعمق تسلسل معزز.
ببساطة، يمكن لـ RDA تقليل عمق التسلسل، بحيث بالإضافة إلى إكمال مهمة استرداد القناع التقليدية، يمكن للنموذج أيضًا استرداد معلومات التعبير الجيني للخلايا عالية الجودة من الخلايا ذات الجودة المنخفضة أثناء مرحلة ما قبل التدريب.
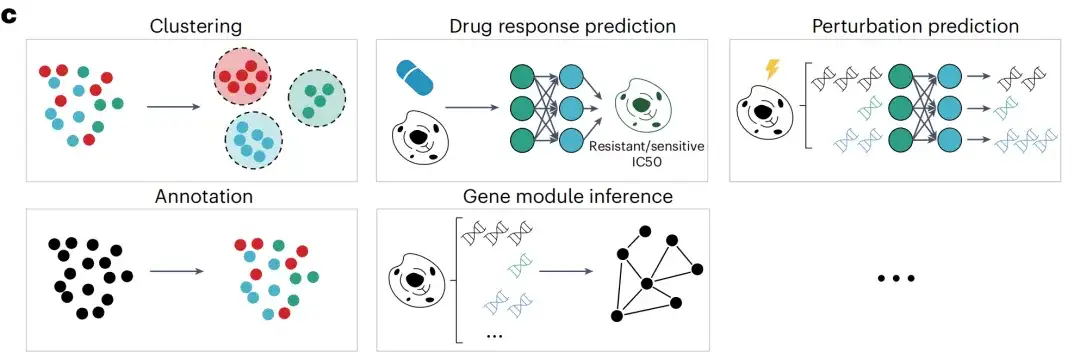
بعد التدريب المسبق، قام الباحثون بتطبيق نموذج scFoundation بشكل أكبر على مهام متعددة لاحقة. يتم تلخيص مخرجات مشفرات scFoundation في متجهات على مستوى الخلية لاستخدامها في المهام على مستوى الخلية بما في ذلك التجميع (داخل مجموعات البيانات وعبرها)، والتنبؤ باستجابة الدواء على مستوى الدفعة والخلية الفردية، وشرح نوع الخلية. إن مخرجات فك تشفير scFoundation عبارة عن متجه سياق على مستوى الجين، والذي يتم استخدامه للمهام على مستوى الجين مثل التنبؤ بالاضطراب واستنتاج وحدة الجين.
نتائج البحث: نماذج scFoundation تتمتع بأداء متفوق
في التطبيقات الفعلية، يدعم نموذج scFoundation وضعين: "جاهز للاستخدام" و"الضبط الدقيق".في الوضع "الجاهز للاستخدام"، وبفضل مهام التدريب المسبق الفريدة، يمكن استخدام النموذج بشكل مباشر لتحسين جودة بيانات الخلية ويمكنه تحقيق نفس النتائج أو نتائج أفضل من الطرق الحالية دون تعديل إضافي. بالإضافة إلى ذلك، يمكن للمستخدمين استخدام scFoundation لاستخراج تمثيلات مدربة مسبقًا للخلايا، والتي يمكن استخدامها لتحديد وحدات الجينات الخاصة بنوع الخلية وعوامل النسخ ويمكن استخدامها على نطاق واسع في المهام اللاحقة.
نموذج تعزيز متسلسل عميق قابل للتطوير وخالي من الضبط الدقيق
قام الباحثون بتدريب ثلاثة نماذج باستخدام معلمات 3M و10M و100M على التوالي، وسجلوا خسائرهم على مجموعة بيانات التحقق.
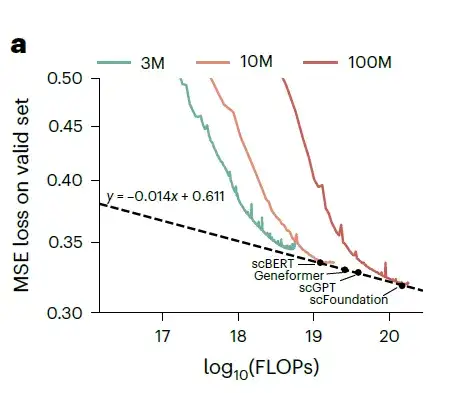
خسارة التدريب تحت أحجام معلمات مختلفة وFLOPs. يمثل المنحنى الأخضر نموذج 3M، ويمثل المنحنى البرتقالي نموذج 10M، ويمثل المنحنى الأحمر نموذج 100M.
مع زيادة معلمات النموذج وعمليات النقطة العائمة (FLOPs)، يظهر الخسارة في مجموعة بيانات التحقق انخفاضًا وفقًا لقانون القوة. قام الباحثون بعد ذلك بتقدير أداء نماذج بنية xTrimoGene بأحجام مختلفة وقارنوها مع scVI. كما هو موضح في الشكل أعلاه،حقق نموذج scFoundation الذي يحتوي على 100 مليون معلمة أفضل أداء بين جميع النماذج.قام الباحثون بتقييم النماذج الثلاثة بشكل أكبر في مهمة شرح نوع الخلية ولاحظوا أن الأداء تحسن مع زيادة حجم النموذج.
قام الباحثون بتقييم هذه القدرة على مجموعة بيانات اختبار مستقلة مكونة من 10000 خلية تم أخذ عينات عشوائية منها من مجموعة بيانات التحقق، عن طريق تقليص العدد الإجمالي إلى 1%، و5%، و10%، و20% من البيانات الأصلية، مما أدى إلى إنشاء أربع مجموعات بيانات مع اختلافات مختلفة في العدد الإجمالي. بالنسبة لكل مجموعة بيانات، تم قياس متوسط الخطأ المطلق (MAE)، ومتوسط الخطأ النسبي (MRE)، ومعامل ارتباط بيرسون (PCC) بين القيم المتوقعة والتعبير الجيني الفعلي غير الصفري باستخدام scFoundation غير المضبوط.
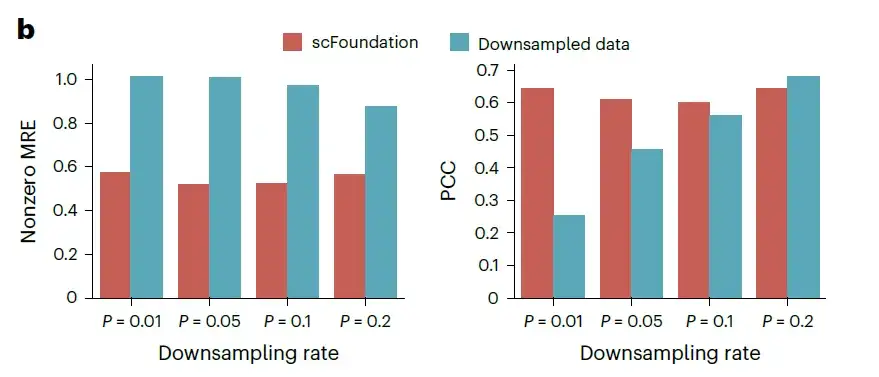
تقييم أداء تعزيز عمق القراءة على مجموعات البيانات غير المعروفة تم استخدام MRE و PCC لتقييم أداء التعبير الجيني المسترد، حيث يشير انخفاض MRE وارتفاع PCC إلى أداء أفضل.
كما هو موضح في الشكل أعلاه، حتى عندما يكون معدل أخذ العينات أقل من 10%، يتم تقليل MAE وMRE لـ scFoundation بشكل كبير إلى النصف.تظهر هذه النتائج قدرة scFoundation على تعزيز التعبير الجيني عند أعداد إجمالية منخفضة للغاية.
المهمة التالية - مهمة التنبؤ باستجابة دواء السرطان
تهدف استجابات أدوية السرطان (CDRs) إلى دراسة استجابات الخلايا السرطانية للتدخل الدوائي، والتنبؤ الحسابي باستجابات أدوية السرطان (CDRs) أمر بالغ الأهمية لتوجيه تصميم الأدوية المضادة للسرطان وفهم بيولوجيا السرطان. في هذه الدراسة، قام الباحثون بدمج scFoundation مع طريقة التنبؤ بـ CDR DeepCDR للتنبؤ بقيمة التركيز المثبط الأقصى النصف IC50 للأدوية في بيانات خطوط الخلايا المتعددة للتحقق مما إذا كان scFoundation يمكن أن يوفر معلومات تضمين مفيدة لبيانات التعبير الجيني الشاملة بناءً على تدريب الخلية الفردية.
قام الباحثون بتقييم أداء النتائج المستندة إلى scFoundation مقابل النتائج المستندة إلى التعبير الجيني عبر العديد من الأدوية وخطوط الخلايا السرطانية.وأظهرت النتائج أن معظم الأدوية وجميع أنواع السرطان حققت معاملات ارتباط بيرسون (PCC) أعلى باستخدام تضمين scFoundation.كما هو موضح في الشكل التالي:
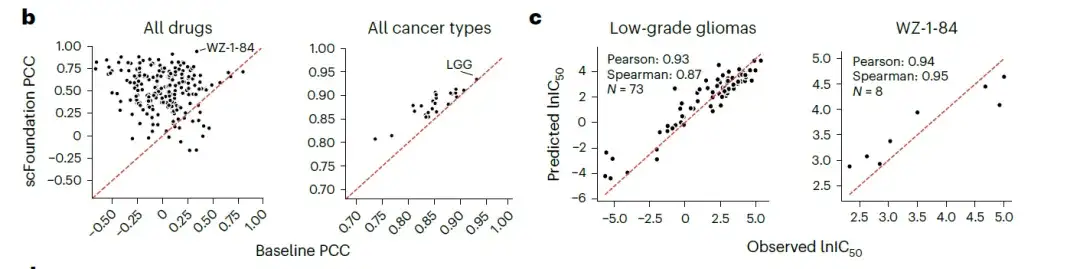
- ملاحظة: معامل ارتباط بيرسون هو إحصائية تقيس قوة العلاقة الخطية بين المتغيرات، ويتراوح نطاق قيمته بين -1 و1. إذا كان معامل الارتباط قريبًا من 1، فهذا يشير إلى وجود علاقة خطية موجبة تمامًا بين المتغيرين؛ إذا كان قريبًا من -1، فهذا يشير إلى وجود علاقة خطية سلبية تمامًا؛ إذا كان قريبًا من 0، فهذا يشير إلى عدم وجود علاقة خطية بين المتغيرين.
وهذا يدل على أنعلى الرغم من أن scFoundation مدرب مسبقًا على بيانات النسخ الجيني للخلية الفردية، إلا أنه من الممكن نقل العلاقات الجينية المكتسبة إلى بيانات التعبير العالمية.يقوم بإنشاء متجه مضغوط، مما يسهل التنبؤات IC50 الأكثر دقة. ولذلك، فإن مؤسسة scFoundation لديها القدرة على توسيع فهمنا لاستجابات الأدوية في بيولوجيا السرطان وتوجيه تصميم علاجات أكثر فعالية لمكافحة السرطان.
المهمة التالية - مهمة تصنيف استجابة الدواء في الخلية الواحدة
إن استنتاج حساسية الدواء على مستوى الخلية الواحدة يمكن أن يساعد في تحديد أنواع فرعية محددة من الخلايا التي تظهر أنماط مقاومة مميزة للأدوية، مما يوفر رؤى قيمة حول الآليات الأساسية والنهج العلاجية الجديدة. ولذلك، قام الباحثون بتطبيق scFoundation على المهمة الحاسمة المتمثلة في تصنيف استجابة الخلية الواحدة للأدوية، استنادًا إلى نموذج لاحق يسمى SCAD.
ركز الباحثون على أربعة أدوية (سورافينيب، NVP-TAE684، PLX4720 وإيتوبوسيد) التي أظهرت قيم AUC (مساحة تحت المنحنى) أقل في الدراسات الأصلية. تمت مقارنة النموذج القائم على scFoundation مع نموذج SCAD الأساسي الذي استخدم جميع قيم التعبير الجيني كمدخلات. وأظهرت النتائج أن النموذج القائم على scFoundation حقق درجات أعلى في قيم AUC لجميع الأدوية، وخاصة بالنسبة لـ NVP-TAE684 و sorafenib، مع زيادة قيم AUC بأكثر من 0.2، كما هو موضح في الشكل أدناه.
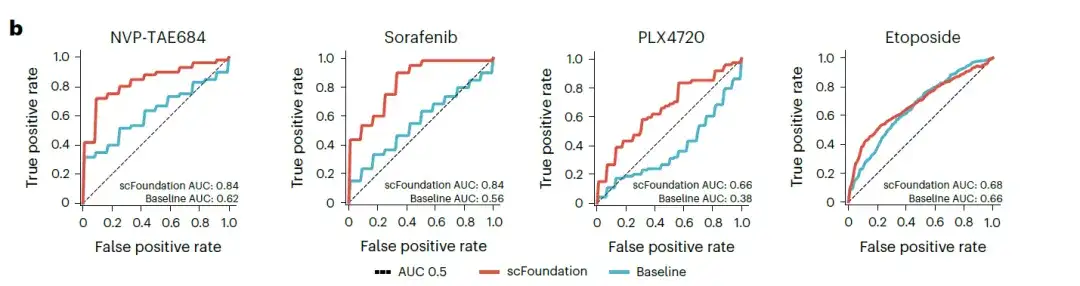
ملاحظة: يمكن استخدام AUC لقياس أداء النموذج. يتراوح نطاق قيمة AUC بين 0 و1. وكلما كانت القيمة أكبر، كان أداء تصنيف النموذج أفضل.
تؤكد هذه النتائج إمكانية استخدام تضمين scFoundation لالتقاط إشارات المؤشرات الحيوية لحساسية الأدوية.
المهمة التالية - مهمة التنبؤ باضطراب الخلية
يعد فهم الاستجابات الخلوية للاضطرابات أمرًا بالغ الأهمية للتطبيقات الطبية الحيوية وتصميم الأدوية، حيث يساعد في تحديد التفاعلات بين الجينات وأهداف الأدوية المحتملة عبر أنواع مختلفة من الخلايا. قام الباحثون بدمج scFoundation مع نموذج متقدم، GEARS، للتنبؤ باستجابات الاضطراب بدقة الخلية الفردية وقاموا بحساب متوسط الخطأ التربيعي (MSE) لأفضل 20 جينًا معبرًا عنه تفاضليًا (DE) مع ملفات تعريف تعبير جيني مختلفة بشكل كبير قبل وبعد كمعيار للتقييم.
وتظهر النتائج أنوبالمقارنة مع نموذج GEARS الأساسي الأصلي، حقق النموذج القائم على scFoundation قيم MSE أقل.يوضح الشكل أدناه تغييرات التعبير عن أفضل 20 جينًا في اضطراب الجين المزدوج ETS2 + CEBPE:
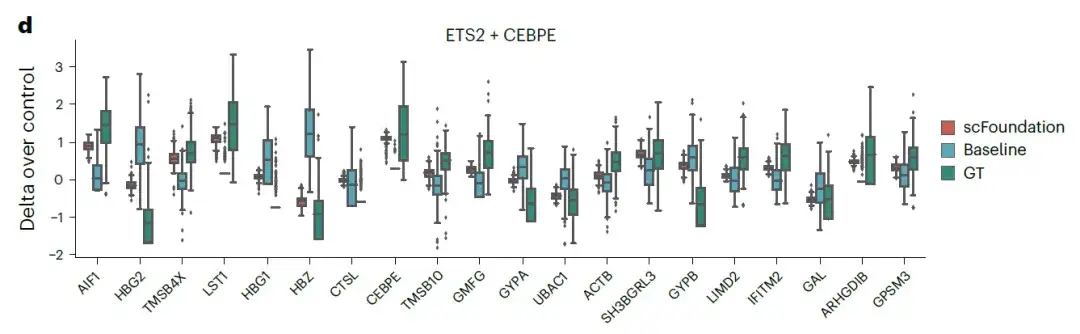
تشير هذه النتائج إلى أنه من خلال استخراج تمثيلات الجينات من الخلايا الفردية لبناء شبكات محددة للتعبير المشترك عن الجينات،نجح scFoundation في التقاط الخصائص الخلوية والجينية في ظل ظروف مختلفة وحسّن بشكل كبير دقة التنبؤات بالاضطرابات الفردية/المزدوجة.
باختصار، يوفر نموذج scFoundation أفكارًا وطرقًا جديدة لإنشاء بنية النموذج وإطار التدريب ونظام تطبيق العرض التوضيحي النهائي لنماذج التدريب المسبق للخلايا الكبيرة، ويوفر وظائف أساسية لتعلم المهام الطبية الحيوية، ويوسع حدود النماذج الأساسية في مجال الخلية الواحدة.
استكشاف نماذج علوم الحياة الكبيرة ذات الأداء الأفضل
قال ديميس هاسابيس، الرئيس التنفيذي ومؤسس شركة DeepMind، وهي شركة رائدة عالميًا في مجال الذكاء الاصطناعي، ذات مرة:على المستوى الأساسي، يُمكن اعتبار علم الأحياء نظامًا مُعقّدًا وديناميكيًا للغاية لمعالجة المعلومات. وكما أثبتت الرياضيات أنها اللغة الوصفية الأنسب للفيزياء، فقد يكون علم الأحياء مجالًا مثاليًا لتطبيقات الذكاء الاصطناعي.
ومع ذلك، تتطلب أساليب الذكاء الاصطناعي التقليدية كميات كبيرة من البيانات المصنفة للتوصل إلى تنبؤات دقيقة. ولكن في علوم الحياة، غالباً ما تكون البيانات المصنفة عالية الجودة قليلة. إن الرغبة في بناء نماذج مهام لاحقة أكثر دقة استنادًا إلى بيانات أقل تعني أن النموذج الأساسي الأساسي يحتاج إلى تمثيل أفضل أو قدرات عامة. ولذلك، بدأ عدد متزايد من الباحثين في العمل على تصميم نماذج كلية عمودية أفضل في المجال البيولوجي.
مايو 2023أصدر فريق بحثي من جامعة تورنتو أول نموذج لغوي واسع النطاق يعتمد على علم الأحياء أحادي الخلية، scGPT.تم تدريب النموذج مسبقًا على أكثر من 10 ملايين خلية، مما يتيح نقل التعلم عبر المهام المختلفة اللاحقة. في يوليو من نفس العام، حاول الفريق تحديث scGPT من خلال إنشاء تدريب مسبق على أكثر من 33 مليون خلية. أظهرت النتائج أن scGPT يمكنه استخراج رؤى بيولوجية رئيسية حول الجينات والخلايا بشكل فعال وتحقيق أداء متقدم في العديد من المهام اللاحقة، بما في ذلك التكامل متعدد الدفعات، والتكامل متعدد الأوميكس، وشرح نوع الخلية، والتنبؤ بالاضطرابات الجينية، واستنتاج شبكة الجينات.
نُشرت الدراسة، التي تحمل عنوان "scGPT: نحو بناء نموذج أساسي لجينومات الخلية الواحدة المتعددة باستخدام الذكاء الاصطناعي التوليدي"، في مجلة Nature Methods.
* رابط الورقة:https://www.nature.com/articles/s41592-024-02201-0
سبتمبر 2023نجح اتحاد Xcompass، وهو فريق بحثي متعدد التخصصات من الأكاديمية الصينية للعلوم، في بناء أول نموذج أساسي للحياة بين الأنواع في العالم - GeneCompass.يدمج النموذج بيانات النسخ لأكثر من 126 مليون خلية مفردة من البشر والفئران، ويدمج أربعة أنواع من المعرفة السابقة بما في ذلك تسلسلات المحفز وعلاقات التعبير المشترك للجينات، ويحتوي على 130 مليون معلمة نموذجية أساسية، مما يحقق التعلم الشامل وفهم قوانين تنظيم التعبير الجيني، مع دعم التنبؤ بتغيرات حالة الخلية والتحليل الدقيق لمختلف العمليات الحيوية.

نُشرت الدراسة على bioRxiv تحت عنوان "GeneCompass: فك رموز آليات تنظيم الجينات العالمية باستخدام نموذج الأساس عبر الأنواع المستند إلى المعرفة".
في أكتوبر 2023، أعلنت شركة الأدوية العالمية العملاقة سانوفي عن شراكة استراتيجية واسعة النطاق مع شركة BioMap BioScience. سيعمل الطرفان بشكل مشترك على تطوير نماذج متطورة لاكتشاف الأدوية العلاجية الحيوية استنادًا إلى نموذج Life Science AI Foundation Model التابع لشركة BioMap.
بالنظر إلى المستقبل، فإن تطبيق الفهم المعقد وقدرات التوليد المبتكرة للنماذج اللغوية الكبيرة التي تتجاوز الخيال البشري إلى حد كبير على "اللغة الطبيعية" الأكثر تعقيدًا للحياة من شأنه أن يغير حقًا نموذج البحث في علوم الحياة.
مراجع:
1.https://www.jiqizhixin.com/articles/2023-9-29
2.https://www.tsinghua.edu.cn/info/1175/112118.htm
3.https://hope.huanqiu.com/article/4FYZxnpu88J
4.https://www.jiqizhixin.com/articles/2023-7-5-26'








