Command Palette
Search for a command to run...
Korean Version of AlphaFold? Deep Learning Model AlphaPPIMd: for protein-protein Complex Conformation Ensemble Exploration

Proteins play an indispensable role in the arena of life. They are the most active molecules in organisms, participating in cell construction, repair, energy conversion, signal transmission, and countless key biological functions. At the same time, the structure of proteins is closely related to their functions, which are realized through complex interactions with proteins, peptides, nucleotides, and various small molecules. This protein-protein interaction (PPI) is at the core of many biological processes in cells, from cell signaling to immune response to cell cycle regulation.
However, our understanding of the three-dimensional structure of proteins and their interaction properties is still incomplete. Traditional experimental techniques, such as X-ray crystallography and cryo-electron microscopy,Although it can provide high-resolution protein structure information, it is time-consuming and costly.Moreover, there are challenges in analyzing dynamic processes and low-abundance proteins, which greatly limits people's in-depth understanding of protein functions and interaction mechanisms, and thus affects the development of drug design and protein engineering.
In response to this, Dr. Jianmin Wang of Yonsei University and his collaborators combined deep learning with generative AI.Using Transformer-based generative neural network learning to explore the conformational ensemble of protein-protein complexes,The key residues affecting the conformation and dynamics of protein-protein complexes were learned from multiple molecular dynamics (MD) trajectories and provided mechanistic insights into protein-protein binding.

Paper address:
https://doi.org/10.1101/2024.02.24.581708
AlphaPPIMd model: Based on molecular dynamics simulation, with self-attention mechanism as the core
The research team used the barnase-barstar complex trajectory set as a dataset.First, the crystal structure of the barnase-barstar complex was downloaded from the Protein Data Bank (PDB), and the A and D chains were extracted as the initial complex structure by removing the ligands and crystal water. Then, the researchers added the missing hydrogen atoms through the tleap module in AmberTools, neutralized them by adding Na+ and Cl- ions, and solvated them in a 12Å TIP3P water molecule periodic boundary box. Finally, the topology and coordinate files of the system were compiled using the tleap module in AmberTools and the AMBER ff14SB force field.
Subsequently, the research team used the molecular dynamics simulation system to perform 500 typical NVT simulations through the Langevin integrator to minimize the energy. Then, 10,000 steps of NPT simulation were performed at 300K to further reach equilibrium, and the particle network Ewald algorithm was used to calculate the long-range electrostatic interaction, the cutoff value of the direct spatial interaction was set to 1nm, the simulation time step was set to 2fs, and the SHAKE algorithm was set to constrain the length of all bonds involving hydrogen atoms, and then 6 independent 100ns molecular dynamics simulations were performed. All simulations were performed using OpenMM 7.7.
After completing the molecular dynamics simulation,The research team built the AlphaPPIMd model based on Transformer, using a deep generative model to capture protein conformational states that are difficult to analyze using traditional molecular dynamics. The core of the AlphaPPImd framework is the self-attention mechanism, which can capture the key amino acid residue pairs that affect the conformation of protein-protein complexes from MD trajectories.
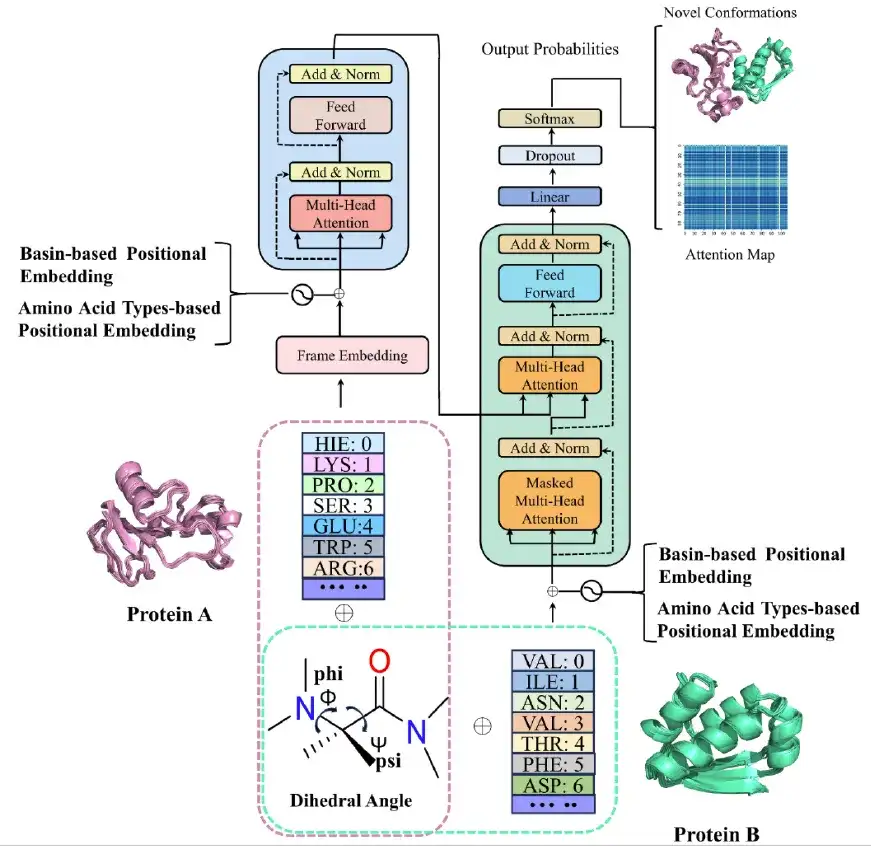
first,The AlphaPPImd framework pre-processes the MD trajectory of the protein-protein complex to obtain the sequence length, sequence composition and amino acid residue type of the two chains, and calculates the Φ, Ψ angles of the selected residues in the trajectory to represent different conformational states. (As shown in the pink and green dotted boxes in the figure above)
Secondly,The researchers fed each frame of the protein-protein complex MD trajectory into the encoder module of AlphaPPImd through an embedding module, which contains a multi-head self-attention mechanism, an attention score, and a feature optimization module. The decoder of AlphaPPImd is used to learn and capture the contributions of residues of different types and positions in protein complexes to the conformation.
at last,The prediction module iteratively generates the ground state for the next frame, and Modeller can reconstruct the conformational model of the protein-protein complex based on the extended ground state encoding trajectory.
The multi-head self-attention layer in the AlphaPPImd decoder module learns the interactions between specific residue pairs, and the attention function can be regarded as a mapping between the query (Q) and the key-value pair (KV) output. AlphaPPImd uses protein complex residue embedding as Q, global protein complex features as K and V, and calculates the attention weight by using Q and K. Its calculation formula is as follows:
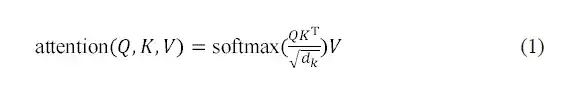
At the same time, the study divided 6 independent 100ns MD trajectories of the barnase-barstar complex into 300 primitives, each consisting of 1,000 frames. The MD trajectories were preprocessed to retain only protein atoms. Each MD run provides a limited set of physical snapshots of the protein-protein complex. Each frame in the trajectory is represented as a Φ,Ψ-encoded ground state. Therefore,The torsion state of a protein-protein complex is reduced to a textual representation,The main minor features of the dynamics are preserved.
Research conclusion: The average training accuracy is as high as 0.995, which can be extended to more protein complexes
The barnase-barstar complex consists of two different chains with a total of 197 residues (barnase chain: 108 residues, barstar chain: 89 residues). The study used the KMeans algorithm to divide the sites into 4 clusters, labeled 0 (purple in the figure below), 1 (dark blue in the figure below), 2 (green in the figure below), 3 (yellow in the figure below), and then recorded and stored the center of mass of each cluster to reconstruct the full-atom model of the barnase-barstar complex from the torsion state encoded in the ground state.
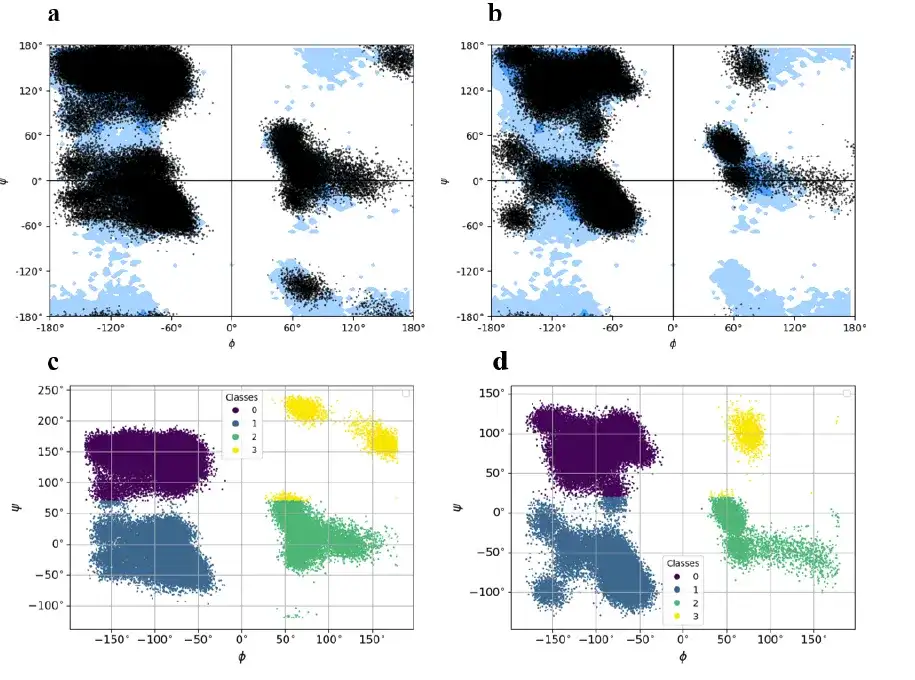
The study converted the trajectory of each frame into a character vector, each of which consisted of 4 symbols corresponding to 4 clusters. Finally, the study performed a similar representation process for all 300 primitives in the MD trajectory dataset of the barnase-barstar complex.
In summary,The barnase-barstar complex is a heterodimer with distinct differences in the ground states of the residues encoded in the two chains.This implies that the barnase-barstar complex differs significantly in generating new ground-state encoding frameworks and in reconstructing conformational models of individual proteins.
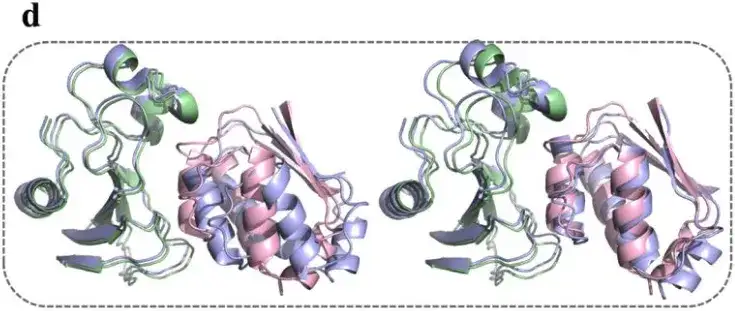
Research shows thatThe average training accuracy of the AlphaPPImd model is 0.995 and the average validation accuracy is 0.999.Although AlphaPPImd quickly achieved stable performance, in order to further improve the Transformer model and enrich the distribution of MD conformations learned by the model, the study used multiple MD trajectories as datasets. For example, the study randomly selected a frame from the trajectory of the test set as input and used the trained AlphaPPImd framework to generate 100 ground state encoding frames.
The results show thatThe model is able to successfully sample and unfold conformations.And the dihedral constraints of Φ and Ψ can be correctly enforced.
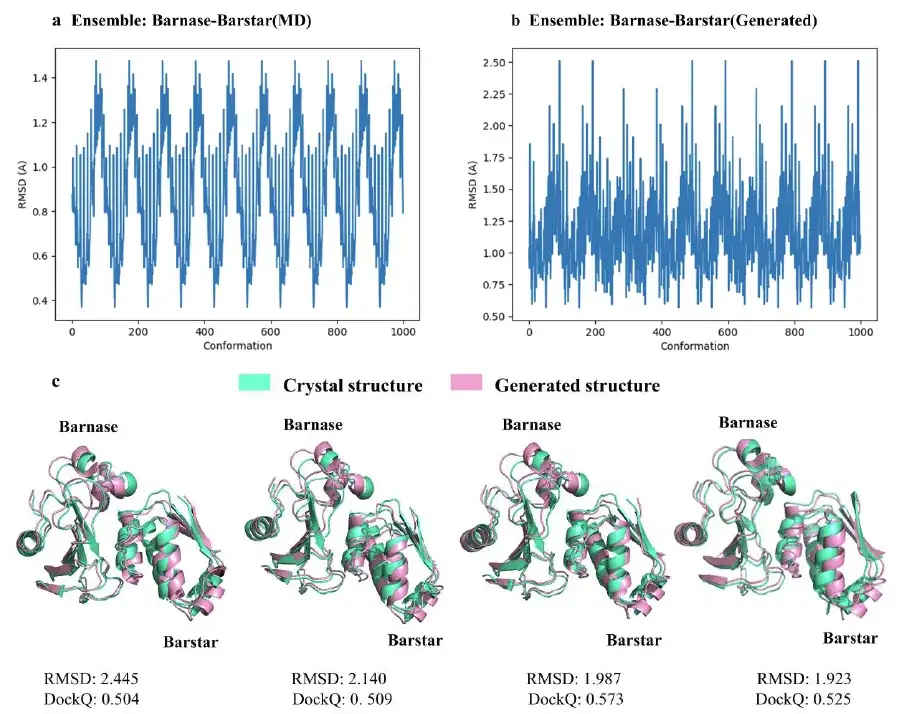
The study also selected four representative conformations with RMSD close to 2Å from the 1,000 barnase-barstar complex conformations generated by the AlphaPPImd model.The protein complex conformation model generated by AlphaPPImd is closer to the reference crystal structure.The accuracy was higher (RMS deviation < 2Å) and the acceptability was higher (DockQ ≥ 0.23).
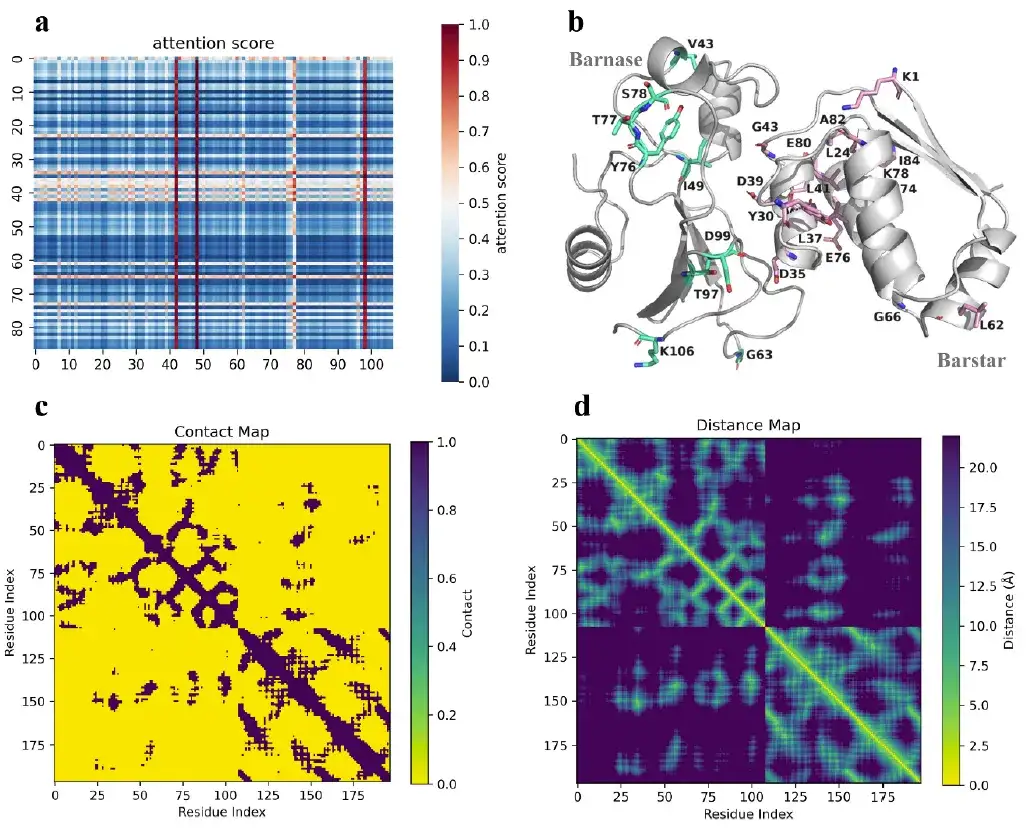
Furthermore, the attention mechanism of AlphaPPImd captures the attention weights between key residues and provides mechanistic insights into protein-protein binding.
Studies have shown that the key residues captured by the AlphaPPImd model are mainly located at the interfaces of protein interactions, loops, and helices, which means thatThe deep generative model captured the key residues that affect the dynamics and conformation of the barnase-barstar complex from the MD trajectory.It can be used to supplement the MD results. At the same time, the key residues captured by the AlphaPPImd model are mainly located in the Mdm2-p53 interaction interface, which also proves that the model can be extended to other protein-protein complexes.
AI protein prediction: from AlphaFold To a hundred schools of thought
As early as 2016, after AlphaGo became famous, the DeepMind team began researching the protein folding problem.
At the end of 2018, in the 13th CASP (Critical Assessment of protein Structure Prediction), AlphaFold ranked first among 98 contestants and accurately predicted the structures of 25 proteins out of 43 proteins. In 2020, AlphaFold 2 was released, achieving high-accuracy predictions of protein monomer structures. In October 2021, DeepMind released an update called AlphaFold-Multimer, which expanded on AlphaFold 2 and can model complexes of multiple proteins. On May 8, 2024, AlphaFold 3 once again amazed the world, extending the scope of prediction from proteins to a wide range of biological molecules.
As early as when AlphaFold 2 was launched, Shi Yigong, an academician of the Chinese Academy of Sciences, told the media: "In my opinion, this is the greatest contribution of artificial intelligence to the field of science. It is also one of the most important scientific breakthroughs made by mankind in the 21st century. It is a very remarkable historical achievement in mankind's scientific exploration of the natural world."
With the example of AlphaFold, the industrial revolution brought about by AI in the field of protein design has quietly arrived.
In 2023,The world's first large-scale AI protein generation model, NewOrigin (Chinese name "Darwin"), was officially unveiled at the World Manufacturing Conference.It is reported that the NewOrigin large model is based on a conditional generation mechanism and combines multi-dimensional feedback mechanisms such as AI, molecular dynamics, quantum computing, and wet experiments. It can generate protein sequences, protein functions, protein knowledge representations and other modal protein contents with high precision, and complete multi-dimensional tasks such as affinity, stability, activity, and expression to meet the needs of real industrial applications.
In 2022, biologists from the University of Washington School of Medicine published two papers in Science, introducing their major discoveries. The researchers said,Using machine learning, protein molecules can be created in seconds.In the past, this time would take several months. Creating proteins that do not exist in nature will help develop vaccines, accelerate research on cancer treatments, develop carbon capture tools, and develop sustainable biomaterials.
There is no doubt that AI protein structure prediction can help us better understand proteins and, in turn, life. However, knowledge and understanding alone are far from enough. In the future, scientists will need to use AI to predict proteins to solve practical problems in the medical field, such as modifying proteins on demand or even designing proteins that do not exist in nature from scratch. The road ahead is long and arduous, and we look forward to AI bringing more surprises in the exploration of life sciences.








