Command Palette
Search for a command to run...
単一細胞トランスクリプトームシーケンス単一サンプルチュートリアル: 品質管理、クラスタリング、(差分)遺伝子表示
単一細胞トランスクリプトームシーケンス解析のチュートリアル
この研究で使用されたサンプルデータは、2024 年 7 月に Nature Medicine に掲載された論文から得たものです。 進行肝細胞癌における個別化ネオアンチゲンワクチンとペムブロリズマブ:第 1/2 相試験 患者番号6の末梢血単核細胞の単一細胞シーケンスデータ(Yarchoan M et al. Nat Med. 2024;30:1044–1053.)。
シングルセルシーケンス入門
単一細胞シーケンシングは、科学者が個々の細胞レベルで遺伝子発現を詳細に分析できるようにする高度なバイオテクノロジーです。このプロセスは通常、最初に組織サンプルを個々の細胞に分離して単一細胞懸濁液を形成することから始まります。次に、油中水技術を使用して、各細胞を個別にカプセル化し、その後の分析中に細胞が独立性を保つようにしました。
単一細胞シーケンスでは、各細胞の mRNA 分子に固有の分子識別子が付けられます。このプロセスは UMI (Unique Molecular Identifier) ラベリングと呼ばれます。同時に、各セルには同じバーコード ラベルが割り当てられ、データ分析段階で各セルのソースを追跡するために使用されます。このようにして、研究者は各細胞の mRNA 情報を正確に測定し、各細胞の遺伝子発現状態を把握することができます。
1. バーコード機能:細胞情報を記録
2. 単一細胞シーケンスにおける UMI の応用: 各 mRNA 分子に固有の識別子を提供することで、遺伝子発現測定の精度が向上するだけでなく、PCR 増幅中に発生する可能性のあるエラーも削減され、シーケンス プロセス全体の信頼性と有効性が向上します。
基本的なプロセス
単一細胞RNAシーケンシング(scRNA-seq)のプロセスには、主に単一細胞の分離と捕捉、細胞溶解、逆転写(RNAをcDNAに変換する)、cDNA増幅、ライブラリの準備が含まれます。
画像出典: Kulkarni, A., et al. (2019). “バルクを超えて: 単一細胞トランスクリプトミクスの方法論と応用のレビュー” カーオピンバイオテクノロジー58: 129-136
ステップの実行
それではチュートリアルの実践部分に入ります。
コンテナを起動してデータをアップロードする
1. コンテナを起動します
コンテナを作成し、rstudio を開いて、package-installed-gpu バージョンを選択し、リソースが割り当てられた後に API アドレスを直接開いて R studio に入ります (この手順では実名認証が必要です)。次の図を参照してください。
- 注: ソフトウェアに入った後のアカウントとパスワードは、rstudio です。
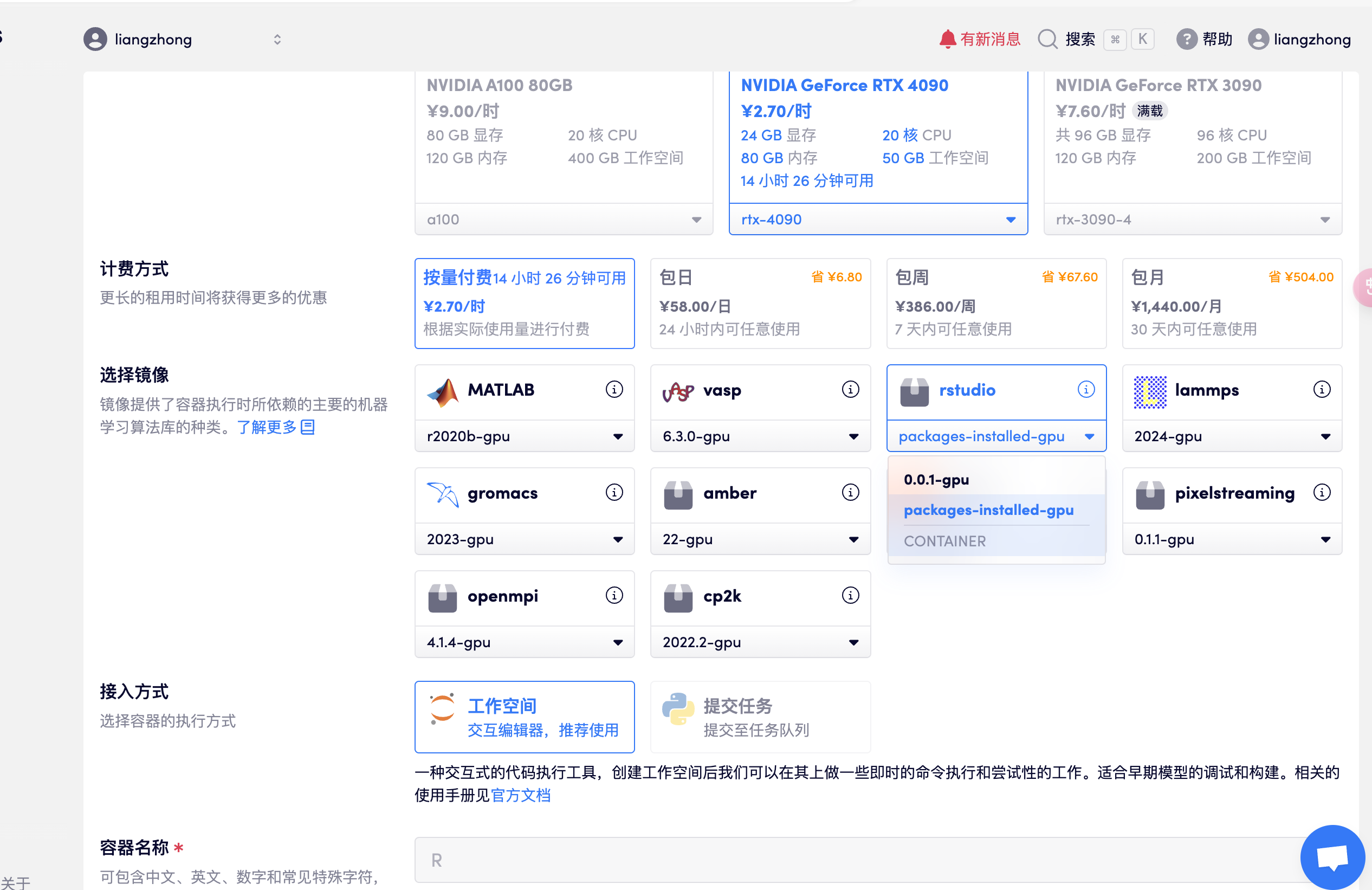
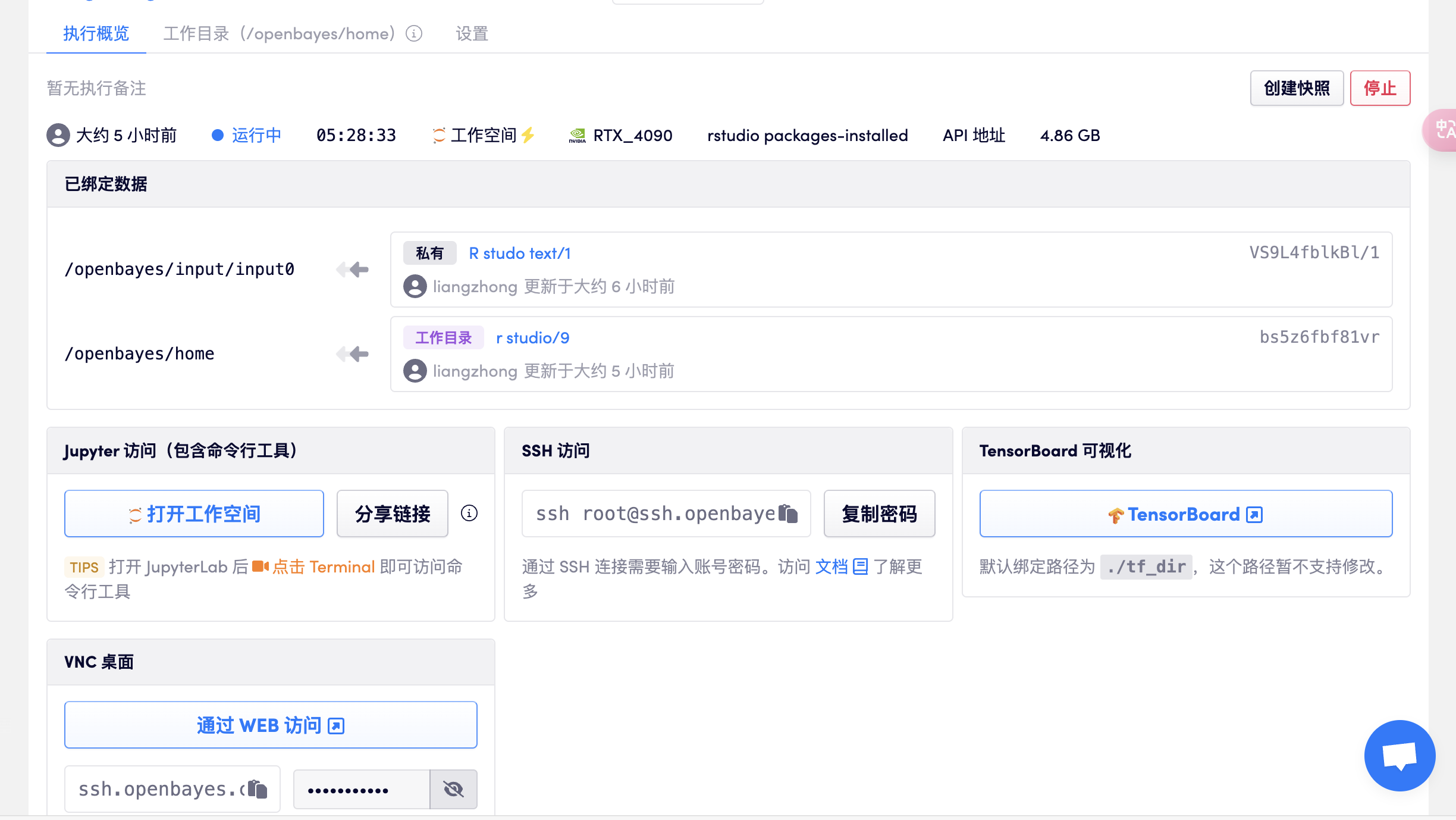
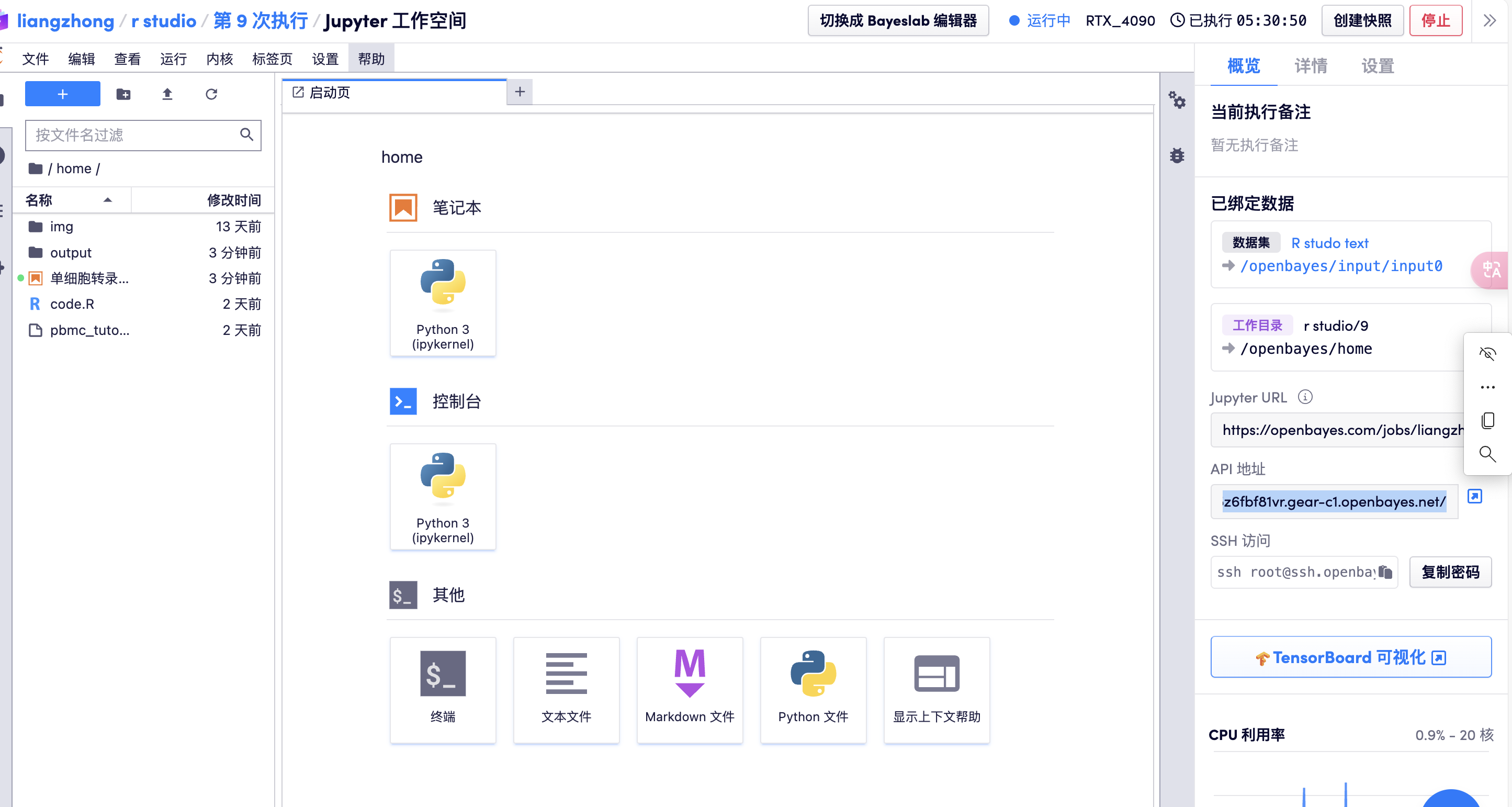
2. データをアップロードする
事前に準備したデータをアップロードします(データは作業ディレクトリのデータフォルダにダウンロードして使用できます)。
(1)イルミナNextSeq 500でシーケンスされた単一細胞の生データ:barcodes.tsv.gz、features.tsv.gz、matrix.mtx.gz
(2)細胞注釈の参照データ:pbmc_multimodal.h5seurat、ref_Human_all.RData
#存放的目录如下
#此处可新建自定义文件夹,指定输入输出文件目录,本教程以 output 为例
rstudio@****(用户名)-bs5z6fbf81vrmain:~/home/output/data$ ls
barcodes.tsv.gz features.tsv.gz matrix.mtx.gz
rstudio@****(用户名)-bs5z6fbf81vr-main:~/home/output/reference_annotation$ ls
pbmc_multimodal.h5seurat ref_Human_all.RData次に、チュートリアルの手順に従って、R スタジオにコードを段階的に入力します。
#报错改为英文
Sys.setenv(LANGUAGE = "en")
options(stringsAsFactors = FALSE)
#清空所有数据
rm(list=ls())
set.seed(100)#!!!设置随机数,保证后续实验的可重复性1. Seuratオブジェクトを設定する
# I. Setup -----------------------------------------------------------------------
# ## a. Load libraries ----------------------------------------------------------------------
```{r libraries}
library(Seurat)
library(SeuratObject)
library(sctransform)
library(patchwork)
library(Seurat)
library(SeuratData)
library(patchwork)
library(SeuratData)
library(ggplot2)
library(tidyverse)
library(glmGamPoi)
library(tools)
library(dplyr)
library(patchwork)2. データの読み込み
# II. Load data-----------------------------------------------------------------------
### a. Load data-----------------------------------------------------------------
getwd()#查看当前工作目录
setwd("~/home/output/results")
#修改工作目录,此处可根据自定义文件夹,指定输入输出文件目录,本教程以 output 为例
pbmc.data <- Read10X(data.dir ="~/home/output/data")#设置 10x 原始数据的工作目录,读取
seurat <- CreateSeuratObject(counts = pbmc.data, project = "pbmc", min.cells = 3, min.features = 200)#创建 seurat 对象
seurat
An object of class Seurat
16356 features across 10066 samples within 1 assay
Active assay: RNA (16356 features, 0 variable features)
1 layer present: counts
#该样本有 10066 个细胞 ,10066 个单细胞的 RNA 表达数据,测序了 16356 个基因
#counts: 通常表示原始的基因表达计数数据
colnames([email protected])
[1] "orig.ident" "nCount_RNA" "nFeature_RNA" "percent.mt" "RNA_snn_res.0.1" "seurat_clusters"
[7] "celltype"
pbmc.data データは下の図に示されており、読み取り数、行名は遺伝子、列名はセル バーコードです。
pbmc.data[c("CD3D", "CD8A", "PTPRC"), 1:30] クラス "dgCMatrix" の 3 x 30 疎行列 [[ 30 の列名 'AAACCTGAGCGAAGGG-1'、'AAACCTGAGGCCCTTG-1'、'AAACCTGAGGGTTTCT-1' を省略 … ]]
1 . 1 . 2 9 . 3 4 . 1 . 2 1 4 CD3D 2 . 3 6 4 1 1 3 . 6 3 4 2 1 2 1 . . 2 2 5 2 3 . 6 . 4 3 CD8A . . . 5 . . . 1 . . . . . . . . . . 1 1 . 6 . . 6 PTPRC 1 2 2 2 3 5 3 3 3 1 3 . 2 4 1 . 1 . 2 9 . 3 4 . 1 2 . 2 1 4
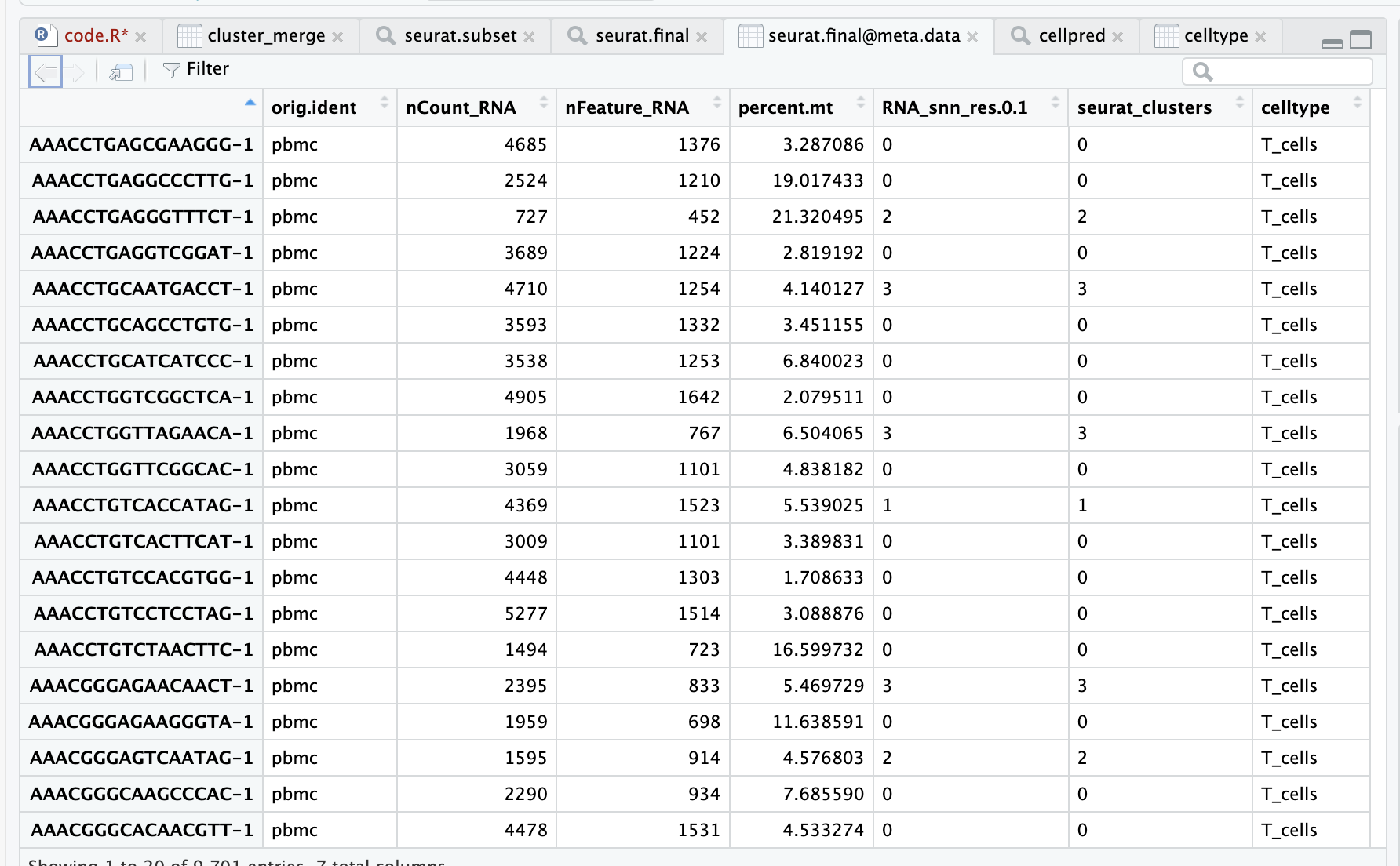
- テーブル情報の説明:
- 行名はバーコードです
- orig.identはサンプル情報です。
- nCount_RNAは総遺伝子発現量である
- nFeature_RNAは遺伝子の数です
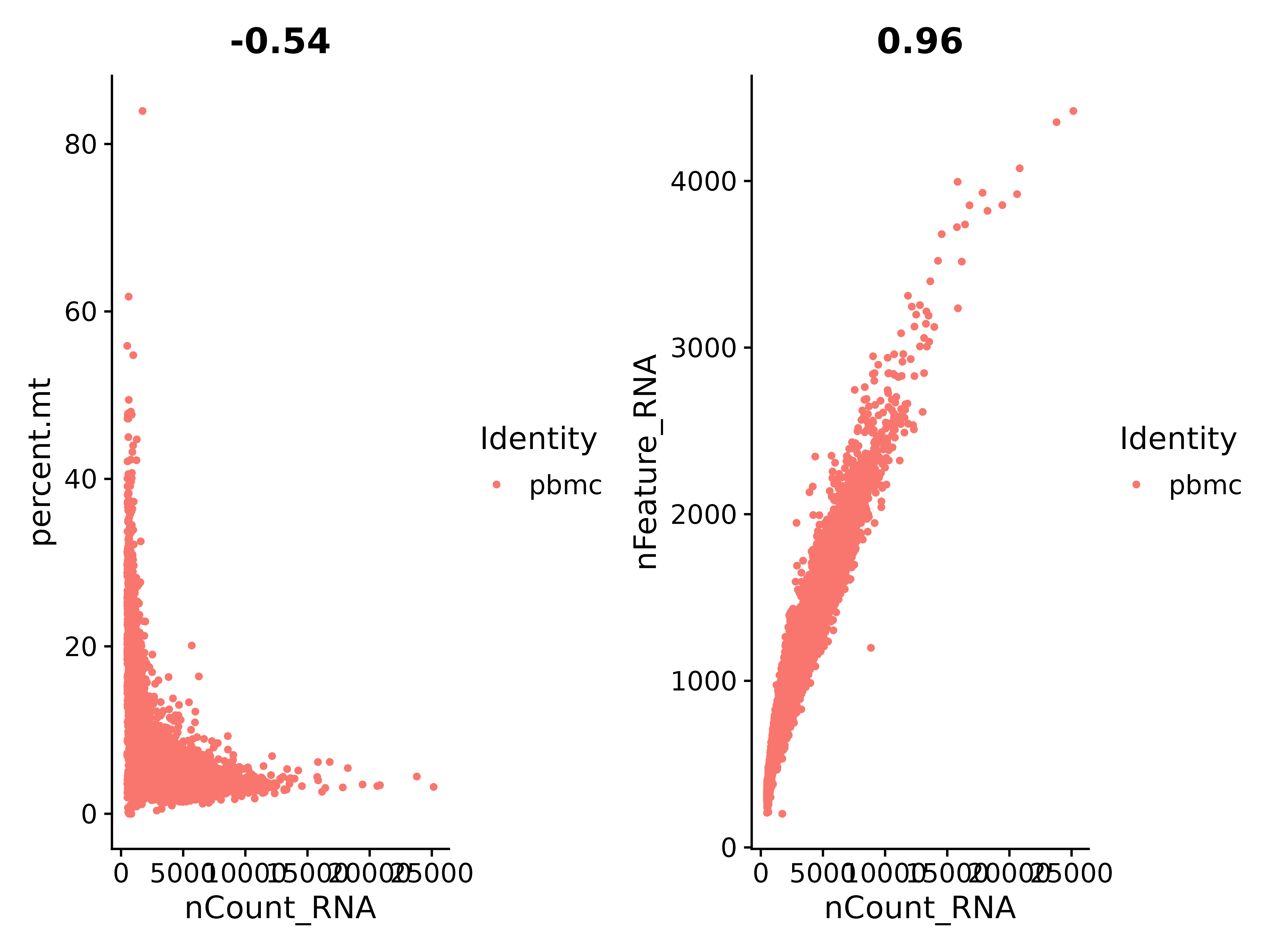
上の図に従って、nCount_RNA と percent.mt (ミトコンドリア遺伝子) の比率を確認してみましょう。
nCount_RNA と nFeature_RNA は係数が 0.96 で正の比率になっており、相関が非常に良好であることがわかります。
ヒント: MT- で始まるすべての遺伝子はミトコンドリア遺伝子です。
3. 標準的な前処理
次の手順は、Seurat における scRNA-seq データの標準的な前処理ワークフローをカバーしています。これらのプロセスには、QC メトリックに基づいたセルの選択とフィルタリング、データの正規化とスケーリング、および非常に変動の大きい特徴の検出が含まれます。
Seurat は、QC メトリックを表示し、ユーザー定義の基準に基づいてセルをフィルタリングできます。
# III. Pre-processing----------------------------------------------------------------
### a. Plots for percent mito, nFeature---------------------------------------------
```{r preprocessing}
# The [[ operator can add columns to object metadata. This is a great place to stash QC stats
seurat <-PercentageFeatureSet(seurat, pattern = "^MT-", col.name = "percent.mt")
# Use FeatureScatter is typically used to visualize feature-feature relationships, but can be used
# for anything calculated by the object, i.e. columns in object metadata, PC scores etc.
plot1 <- FeatureScatter(seurat, feature1 = "nCount_RNA", feature2 = "percent.mt",)
plot2 <- FeatureScatter(seurat, feature1 = "nCount_RNA", feature2 = "nFeature_RNA")
plot1 + plot2
ggsave("质控_1.png", plot1 + plot2, width = 8, height = 6, dpi=600)
#plot3 <- FeatureScatter(seurat, feature1 = "nCount_RNA", feature2 = "percent.ribo")
#plot3
次に、QC メトリックを視覚化し、それを使用してセルをフィルタリングします。
ミトコンドリア遺伝子の割合を見て、バイオリンプロットを使用してQC指標を視覚化する
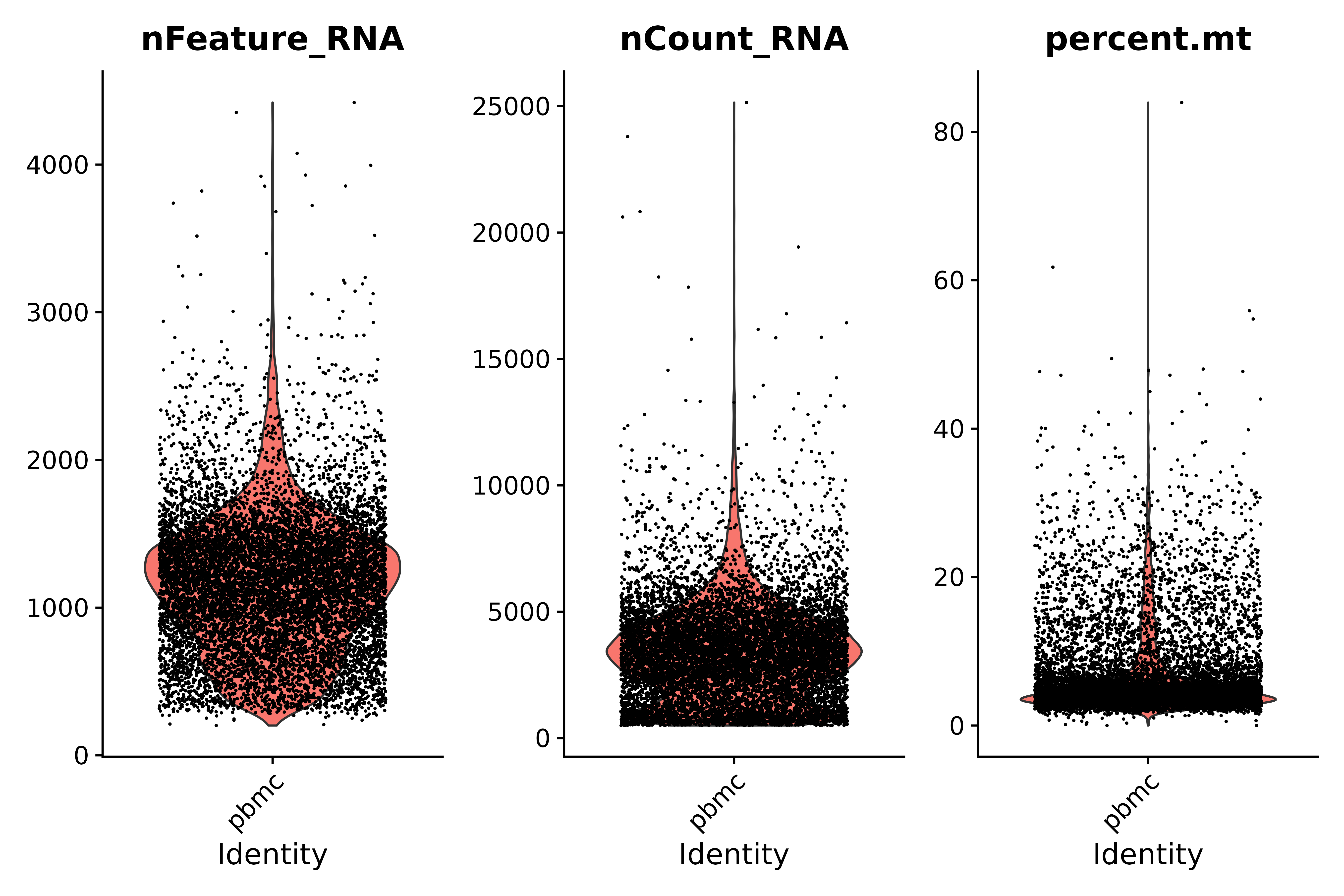
- 品質管理の指示:
- 低品質のセルまたは空の液滴には通常、遺伝子が非常に少なく、nFeature_RNA は 200 を超え、nCount_RNA は 500 を超えます。
- 二倍体または倍数体細胞は異常に高い遺伝子数を示すことがある。私たちのプロットによれば、nFeature_RNA < 2500 & nCount_RNA > 500
- 低品質/死にかけている細胞は通常、広範なミトコンドリア過形成を示し、パーセント.mt < 25
# Visualize QC metrics as a violin plot
vln1 <- VlnPlot(seurat, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"#,"percent.ribo"
),ncol=3)
vln1
ggsave("质控_2.png", vln1, width = 9, height = 6, dpi=600)seurat.subset <- subset(seurat, subset = nFeature_RNA > 200 & nFeature_RNA < 2500 & nCount_RNA > 500 & nCount_RNA < 10000 &percent.mt < 25)
vln3 <- VlnPlot(seurat.subset, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)
vln3
ggsave("质控_3.png", vln3, width = 9, height = 6, dpi=600)
pdf("qc_plots.pdf");plot1 + plot2;vln1;vln3;dev.off()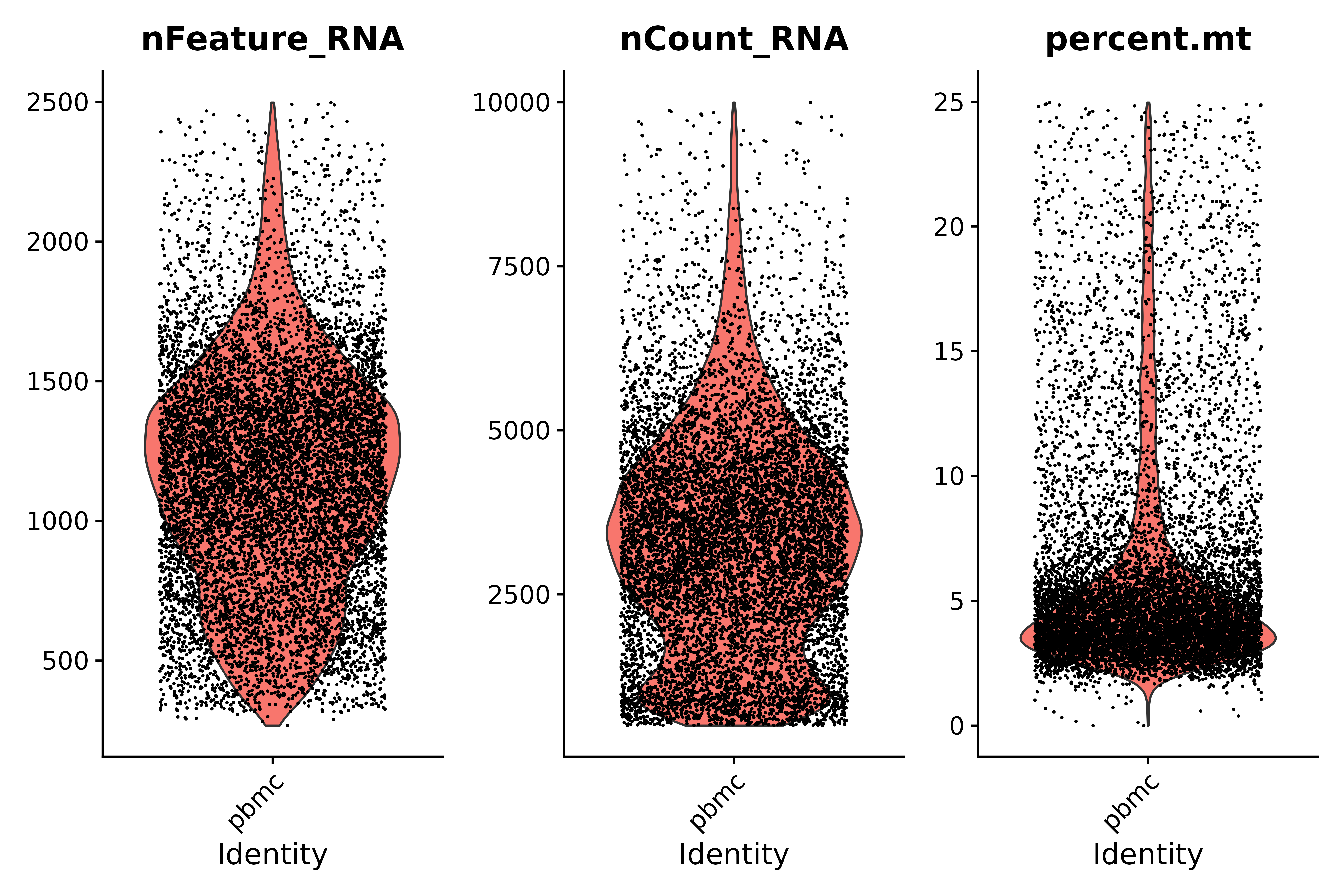
4. データを正規化し、変動の大きい特徴を特定する(特徴選択)
単一細胞トランスクリプトームデータ解析では、まずデータの正規化を実行して、異なる細胞間の遺伝子発現レベルを効果的に比較できるようにします。
1. データの正規化
- データの正規化: まず、各細胞内の特定の遺伝子の発現レベル (カウント) を、細胞の総発現レベル (総カウント) で割ります。このステップは、細胞間のシーケンス深度の差を排除し、異なる細胞の遺伝子発現レベルを同じ基準で比較できるようにするためです。
- スケーリング調整: 正規化された発現値にスケーリング係数 (ここでは 10000) を掛けて、遺伝子発現値を後続の分析に適した大きさに調整します。
- 対数変換: 最後に、調整された発現値に対して対数変換を実行しました。これにより、分散がさらに安定し、高発現遺伝子と低発現遺伝子の違いがより明確になり、後続の分析手順の基礎が築かれました。
- 正規化: 上記の手順により、遺伝子発現値を 10,000 UMI ごとのレベルに正規化しました。これにより、細胞間のシーケンス深度の違いによる影響を排除するのに役立ちます。
2. 次に、突然変異の特徴を検索します。
正規化と標準化を行った後、遺伝子発現の変動性を解析することで、高可変遺伝子を特定しました。これらの遺伝子は細胞によって発現レベルが大きく異なり、ある細胞では高度に発現するが、他の細胞では低レベルで発現する場合があります。
# IV. Normalize Data----------------------------------------------------------------
seurat.subset <- NormalizeData(seurat.subset, normalization.method = "LogNormalize", scale.factor = 10000)
seurat.subset <- FindVariableFeatures(seurat.subset, selection.method = "vst", nfeatures = 2000)
# Identify the 10 most highly variable genes
top10 <- head(VariableFeatures(seurat.subset), 10)
# plot variable features with and without labels
plot3 <- VariableFeaturePlot(seurat.subset)
plot4 <- LabelPoints(plot = plot3, points = top10, repel = TRUE)
ggsave("VariableFeaturePlot_4.png", plot3 + plot4, width = 12, height = 6, dpi=600)
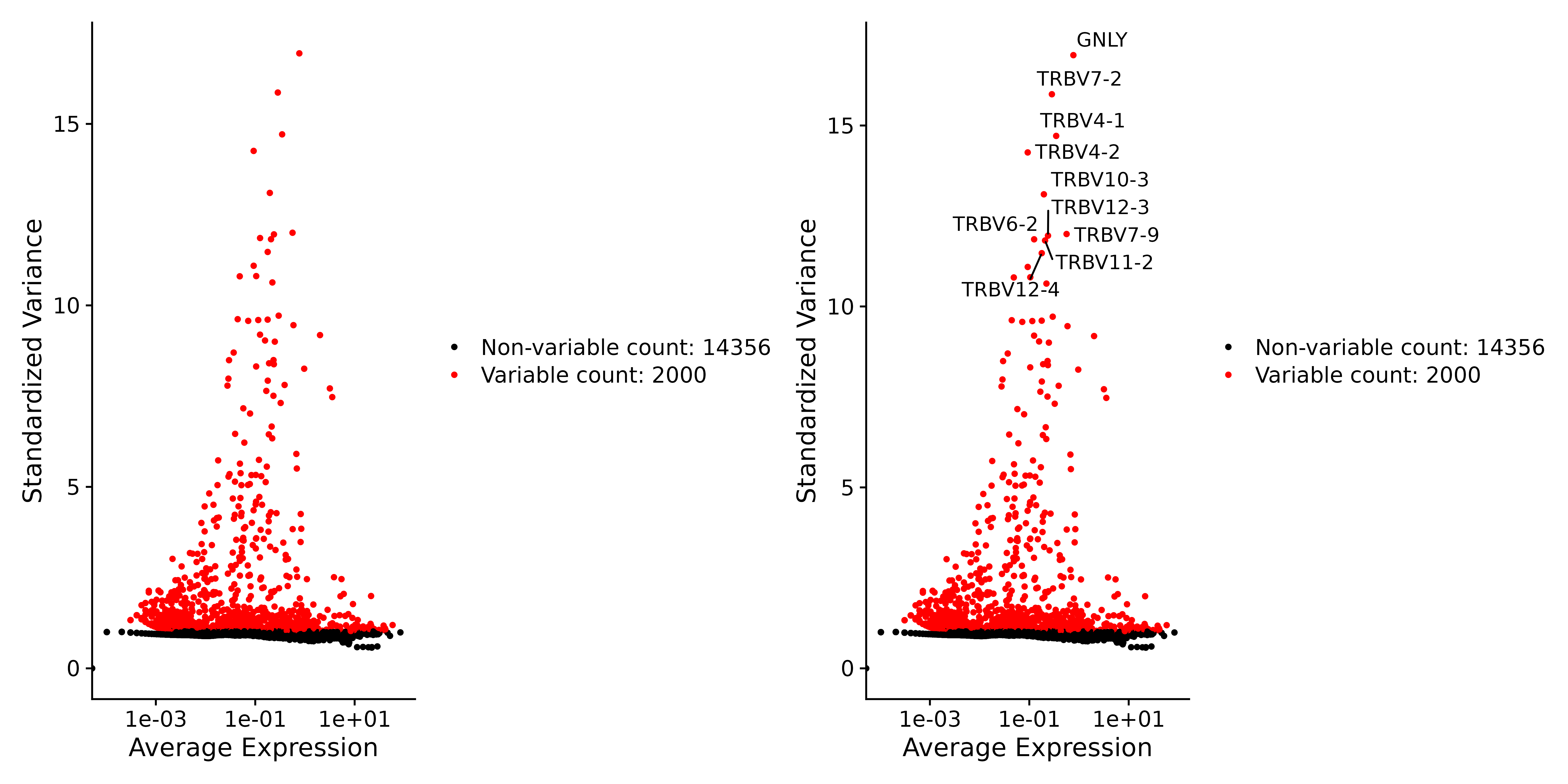
5. データをスケールし、PCAを実行する
単一細胞トランスクリプトームデータ分析では、データスケーリングと PCA により、データの次元を効果的に削減し、細胞間変動の主な原因を抽出し、細胞の異質性と機能を理解するための重要な情報を提供できます。
1. データのスケーリング: データのスケーリングは、遺伝子発現データを標準正規分布、つまりデータの平均が 0 で標準偏差が 1 になるように変換することを目的とした線形変換です。この変換により、異なる遺伝子の発現レベルの違いが軽減され、データがより正規化されます。スケーリングにより、データ内の極端な値や外れ値の影響を軽減し、データ分布をより均一にし、後続の分析ステップでより安定した入力を提供できます。このステップは、PCA (主成分分析) の準備でもあります。PCA は標準化されたデータでより優れたパフォーマンスを発揮するためです。
2. PCAを実行します。
- PCA は、データの投影における最初の最大分散が最初の座標 (第 1 主成分と呼ばれる) 上に、2 番目に大きい分散が 2 番目の座標上に、というように直交軸を変換してデータを新しい座標系に変換する統計手法です。
- 単一細胞トランスクリプトーム解析では、遺伝子発現の違いに基づいて各細胞を分離するために PCA が使用されます。各セルは高次元空間内の点として見ることができ、PCA は可能な限り多くの変動情報を保持しながら、これらの点を低次元空間で再表現します。
- 各主成分 (PCA) は本質的に、セルの「メタ特徴」、つまりセルの変化の主な方向を表します。 PCA の結果では、主成分が前方にあるほど、データ変動におけるその重みが大きくなり、より多くの情報が含まれることを意味します。
- PCA を使用すると、細胞の高次元の発現情報をいくつかの主要な主成分に圧縮することができ、その後のクラスタリング分析、視覚化、またはその他の下流分析に使用できます。
all.genes <- rownames(seurat.subset)
seurat.subset <- ScaleData(seurat.subset, features = all.genes)
seurat.subset <- RunPCA(seurat.subset, features = VariableFeatures(object = seurat))##### Examine and visualize PCA results a few different ways-------------
print(seurat.subset[["pca"]], dims = 1:5, nfeatures = 5)
p1 <- VizDimLoadings(seurat.subset, dims = 1:2, reduction = "pca")
p1
ggsave("VizDimLoadings_5.png", p1, width = 9, height = 6, dpi=600)
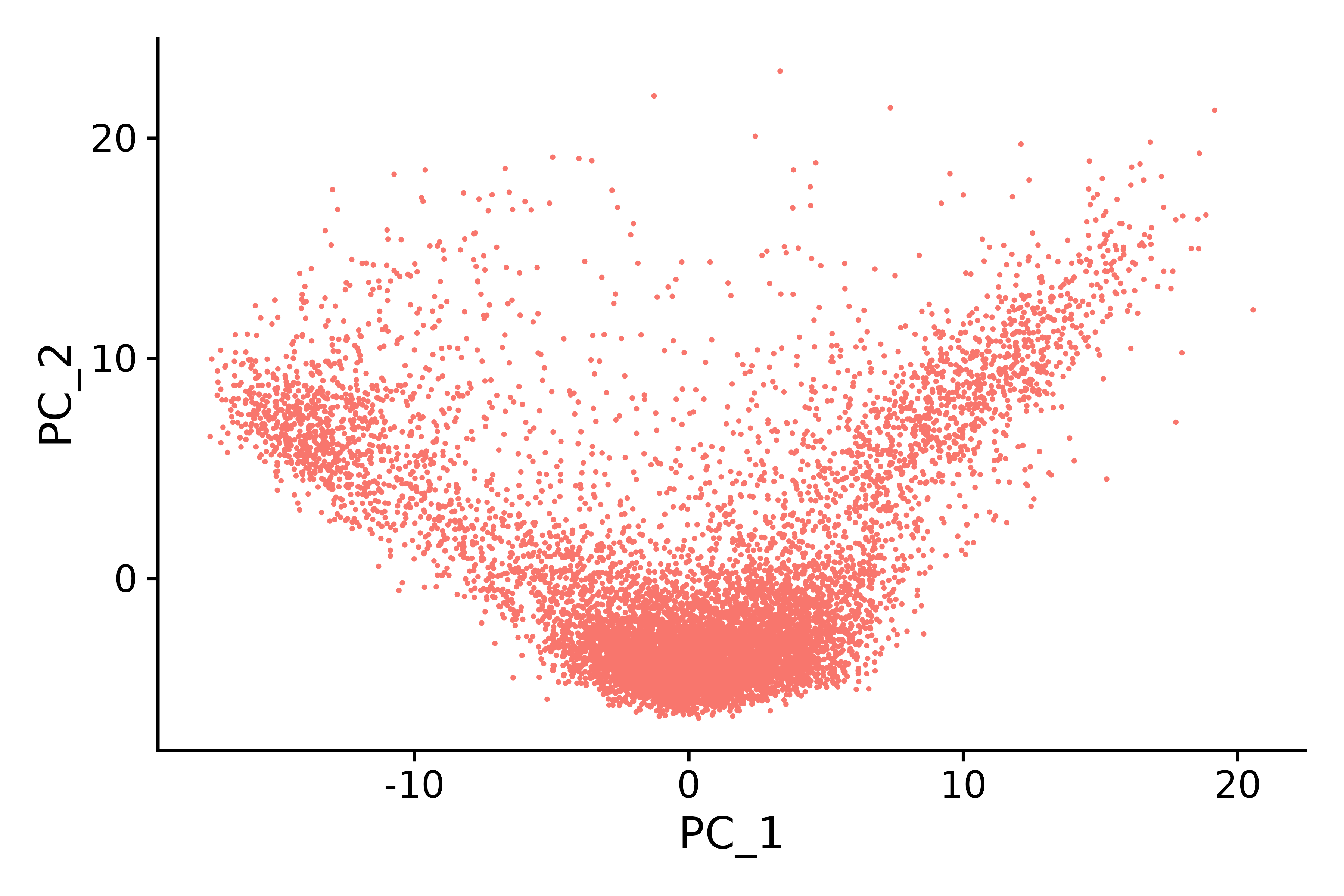
p2 <- DimPlot(seurat.subset, reduction = "pca") + NoLegend()
p2
ggsave("pca_6.png", p2, width = 6, height = 4, dpi=600)
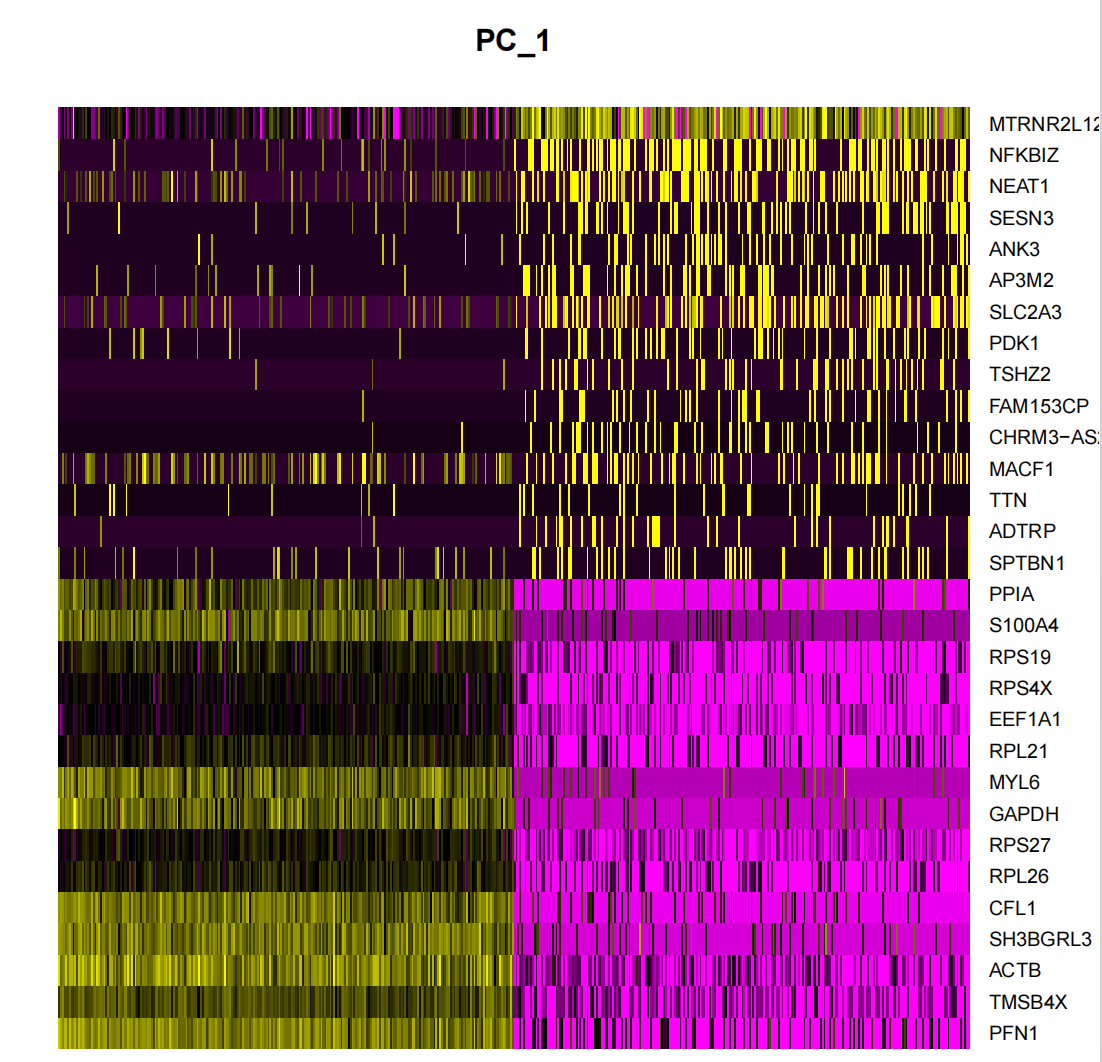
p3 <- DimHeatmap(seurat.subset, dims = 1, cells = 500, balanced = TRUE)
p3
ggsave("pca_1dims.png", p3, width = 6, height = 4, dpi=600)
p4 <- DimHeatmap(seurat.subset, dims = 1:15, cells = 500, balanced = TRUE)
p4
ggsave("pca_15dims.png", p4, width = 10, height = 10, dpi=600)
p5 <- ElbowPlot(seurat.subset)
p5
ggsave("ElbowPlot.png", p5, width = 10, height = 10, dpi=600)
DimHeatmap() は、研究者がデータセット内の異質性の主な原因を簡単に特定して調査できる強力なデータ分析ツールです。主成分分析 (PCA) を実行した後、DimHeatmap() は、その後の詳細な分析に情報を提供するためにどの主成分 (PC) を含める必要があるかを決定するのに特に役立ちます。このプロセスでは、PCA スコアに従ってセルと機能をランク付けし、分析の正確性と関連性を確保します。
さらに、DimHeatmap() を使用すると、ユーザーは特定の数のセルを設定し、データ スペクトルの両端で極端な特性を示す「極端な」セルを具体的にプロットできます。このアプローチでは、データの最も重要な部分に焦点を当て、視覚化プロセス全体を高速化するため、大規模なデータセットを扱う際のプロット効率が大幅に向上します。

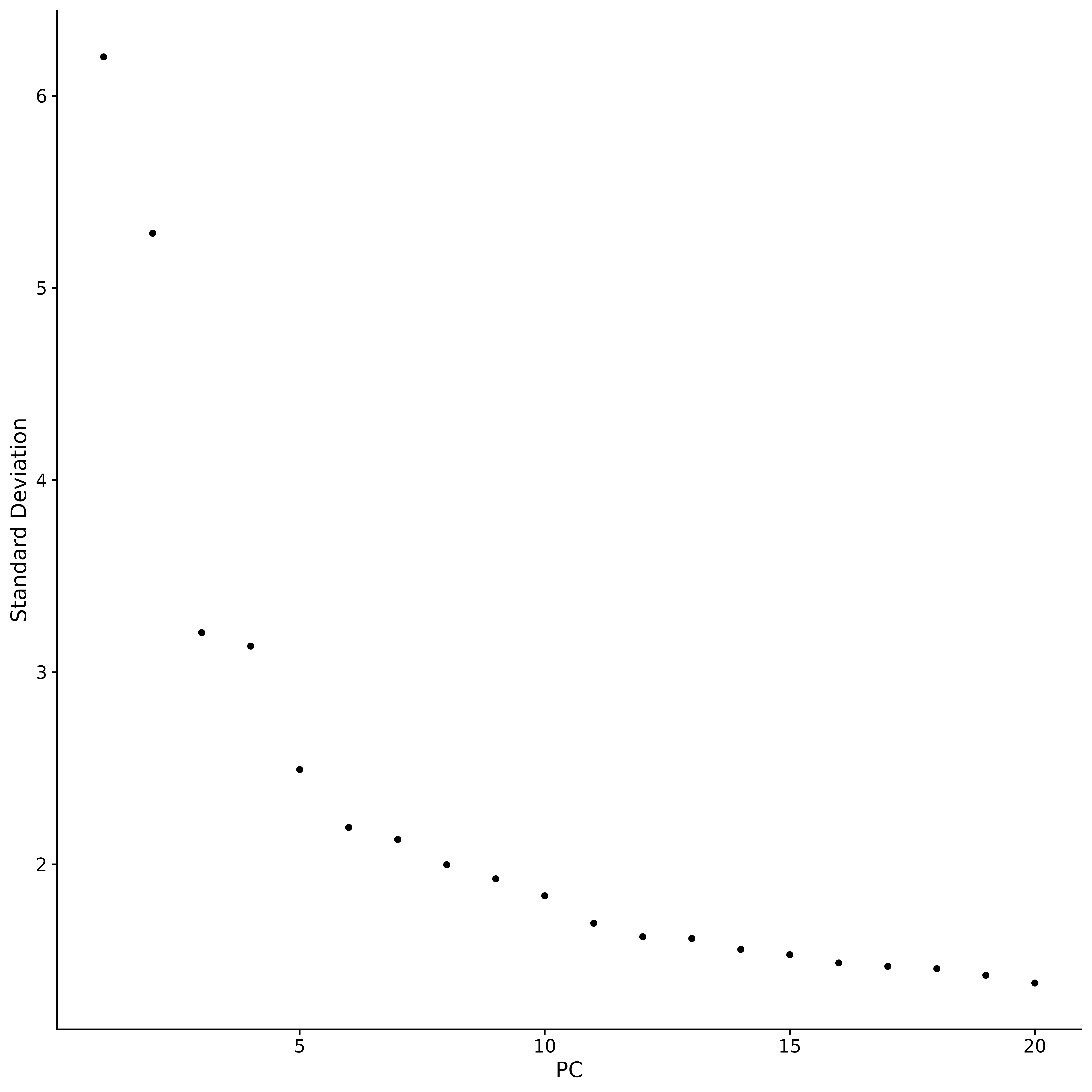
主成分分析 (PCA) では、「エルボー プロット」を使用して、後続の分析で保持する主成分の数を決定するのが一般的です。このプロットは、真のシグナルの大部分が最初の 17 個の主成分に捕捉されていることを示しています。エルボー プロットでは、転換点である「エルボー」を探します。この時点までは、主成分を追加するごとにデータの説明力が大幅に向上しますが、この時点以降は、主成分の追加によるメリットが減少し始めます。これは平衡状態に達したことを示しています。。 エルボープロットの分析結果に基づいて、後続の分析のために最初の 17 個の主成分を選択しました。 主成分を追加してもデータの識別力は大幅に向上せず、計算量が不必要に増加するだけだからです。主成分の数を増やし続けると、データの識別がわずかに改善される可能性がありますが、この改善は重要ではなく、計算コストが大幅に増加します。したがって、17 個の主成分を選択することは、識別と計算効率のバランスをとる合理的な選択です。
pdf("dimreduction_plots.pdf")#保存为 pdf 文件
VizDimLoadings(seurat.subset, dims = 1:2, reduction = "pca")
DimPlot(seurat.subset, reduction = "pca")
DimHeatmap(seurat.subset, dims = 1, cells = 500, balanced = TRUE)
DimHeatmap(seurat.subset, dims = 1:15, cells = 500, balanced = TRUE)
ElbowPlot(seurat.subset)
dev.off()seurat.subset.dimReduction <- seurat.subset
seurat.subset.dimReduction <- FindNeighbors(seurat.subset.dimReduction, reduction="pca", dims = 1:17)
saveRDS(seurat.subset.dimReduction, file = "~/home/output/data/seurat_pre_FindClusters.rds")
# seurat.subset.dimReduction <- readRDS("~/home/output/data/seurat_pre_FindClusters.rds")6. セルのクラスタリング
### 3. Find clusters,聚类
```{r find_clusters}
seurat.final <- FindClusters(object = seurat.subset.dimReduction, resolution = 0.1)
head(Idents(seurat.final), 5)
seurat.final <- RunUMAP(seurat.final, reduction="pca", dims=1:17)
#Visualize with UMAP
p1<- DimPlot(seurat.final, reduction = "umap")
p1
ggsave("find_clusters.png", p1, width = 6, height =5 , dpi=600)
saveRDS(seurat.final, file = "~/home/output/data/seurat_final.rds")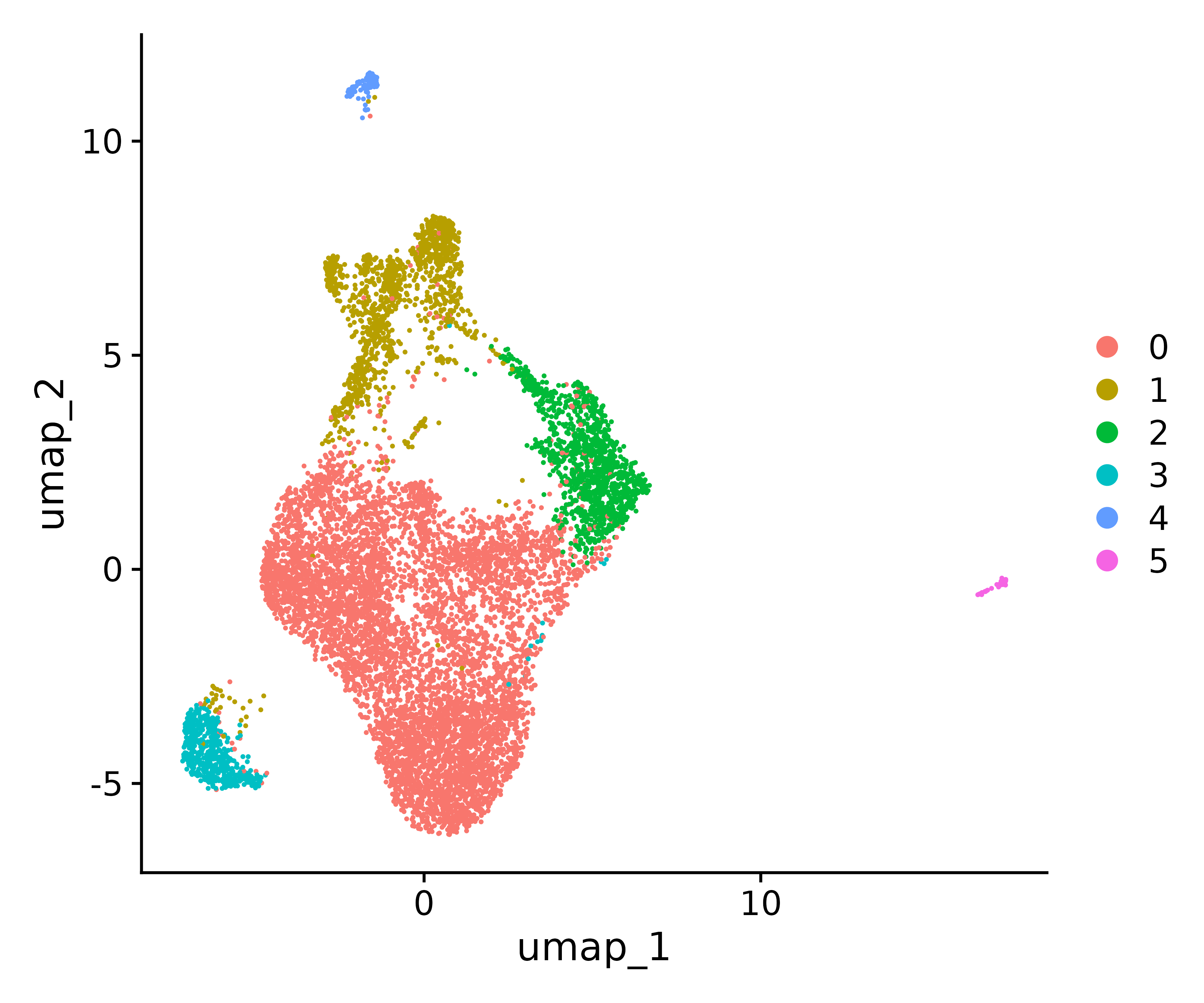
視覚化
異なるクラスター内の遺伝子発現を表示するための一般的なプロットは 3 つあります。
1. ヒートマップ 2. バブルマップ 3. フィーチャプロット
table(Idents(seurat.final))
# find markers between cluster1 and cluster 2
##用来找 cluster 之间的寻找差异表达特征
cluster2.markers <- FindMarkers(seurat.final, ident.1 = 1,ident.2 = 2)
head(cluster2.markers, n = 5)
# find markers for every cluster compared to all remaining cells, report only the positive ones
#找每个 cluster 的 marker 基因
pbmc.markers <- FindAllMarkers(seurat.final, only.pos = TRUE)
ClusterMarker_noRibo <- pbmc.markers[!grepl("^RP[SL]", pbmc.markers$gene, ignore.case = F),]
ClusterMarker_noRibo_noMito <- ClusterMarker_noRibo[!grepl("^MT-", ClusterMarker_noRibo$gene, ignore.case = F),]
###去除核糖体基因和线粒体基因的影响
ClusterMarker_noRibo_noMito %>%
group_by(cluster) %>%
dplyr::filter(avg_log2FC > 1) %>%
slice_head(n = 5) %>%
ungroup() -> top5
###展示每个 cluster 的 top5 的基因
heatmap <- DoHeatmap(seurat.final, features = top5$gene) + NoLegend()
ggsave("heatmap.png", heatmap, width = 10, height =5 , dpi=600) この図はマーカー遺伝子を示すヒートマップである
この図はマーカー遺伝子を示すヒートマップである
g =unique(top5$gene)
##定义 top5 的 gene 列,作为基因集 g,也可以自定义
p <- DotPlot(seurat.final,features=g)+
coord_flip() + # 翻转坐标轴
theme(
panel.grid = element_blank(), # 移除网格线
axis.text.x = element_text(angle = 0, hjust = 0.5, vjust = 0.5) # 旋转 x 轴标签
)
ggsave("allmarker_dotpot.png", p, width = 8, height =7 , dpi=600)
#气泡图展示 marker 基因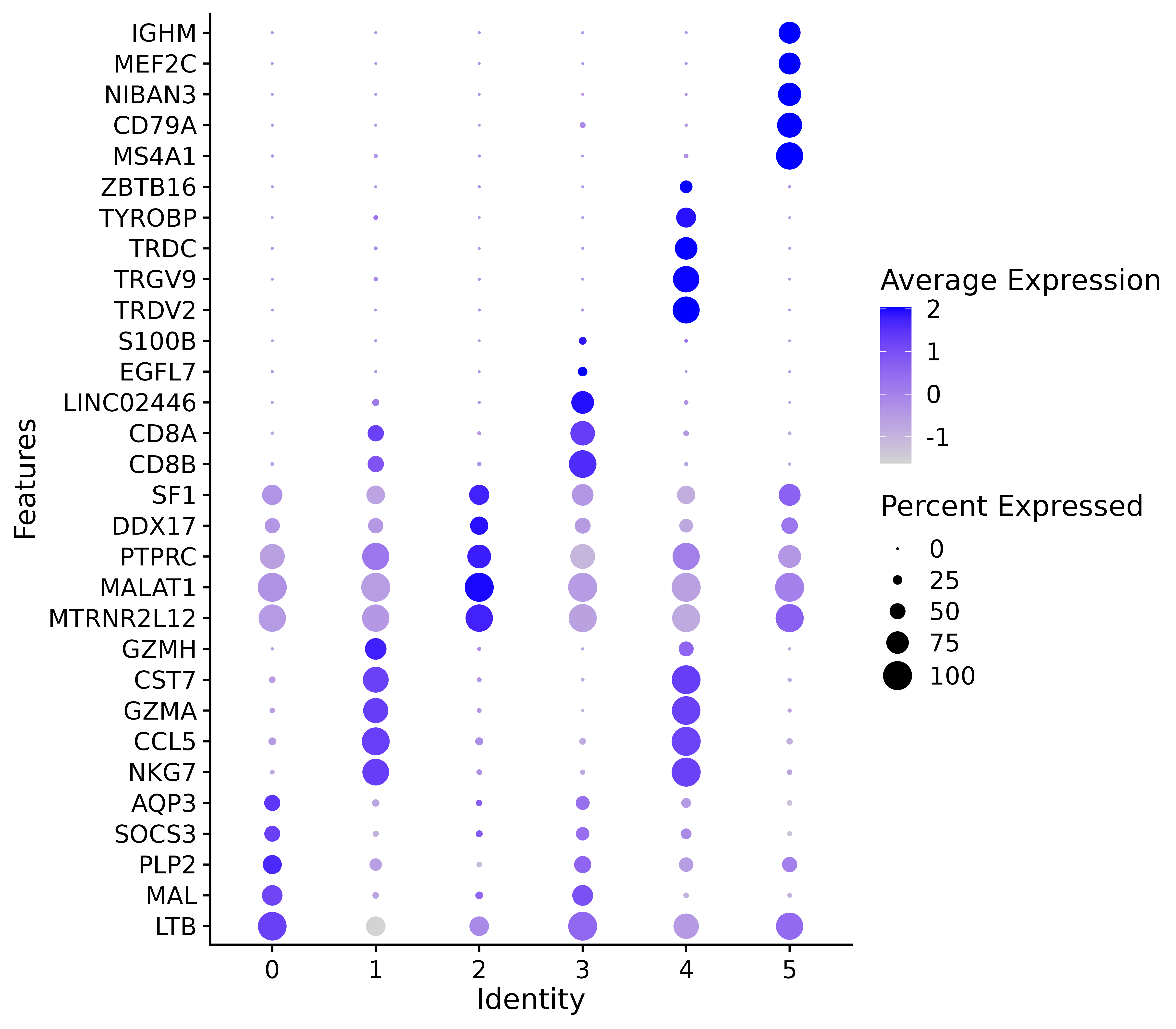
この図はマーカー遺伝子を示すバブルチャートである
# 加载 dplyr 包
library(dplyr)
# 提取 cluster 和 gene 列
cluster_gene_mapping <- top5 %>%
select(cluster, gene)
# 查看结果
head(cluster_gene_mapping,30)
write.csv(cluster_gene_mapping,"cluster_allmarkers_TOP5.csv", row.names = p <- FeaturePlot(seurat.final, features = g)
ggsave("allmarker_featurepot.png", p, width = 15, height =15 , dpi=600)
###查看自定义感兴趣的基因
p2 <- FeaturePlot(seurat.final, features = c("MS4A1", "GNLY", "CD3E", "CD14", "FCER1A", "FCGR3A", "LYZ", "PPBP",
"CD8A"))
ggsave("allmarker_featurepot2.png", p2, width = 15, height =15 , dpi=600)


7. データ分析
このステップでは、単一細胞トランスクリプトーム データ分析で特定のマーカー遺伝子を使用して細胞タイプを決定および定義する方法について説明します。
トップマーカー遺伝子などに基づいて細胞タイプを決定したと仮定すると、アイデンティティタイプを定義できます。
0 1 2 3 4 5 「CD4 T」、「NK」、「Treg」、「CD8 T」、「γδ T細胞」、「B細胞」
(以下のコメントは参考用であり、実際のコメントではありません。)
new.cluster.ids <- c("CD4 T", "NK", "Treg", "CD8 T", "γδ T cell",
"B cell")
names(new.cluster.ids) <- levels(seurat.final)
seurat.final <- RenameIdents(seurat.final, new.cluster.ids)
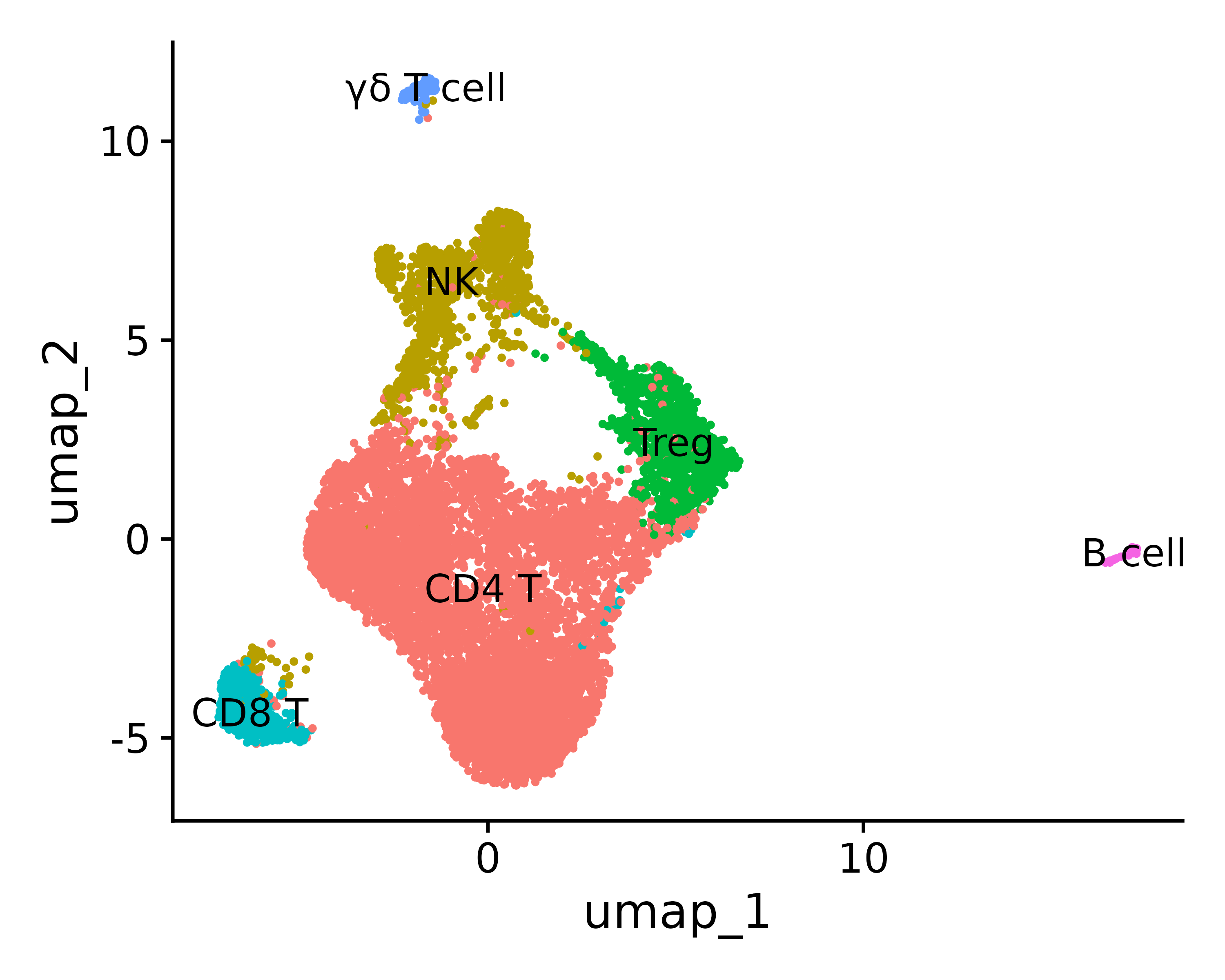
p <- DimPlot(seurat.final, reduction = "umap", label = TRUE, pt.size = 0.5) + NoLegend()
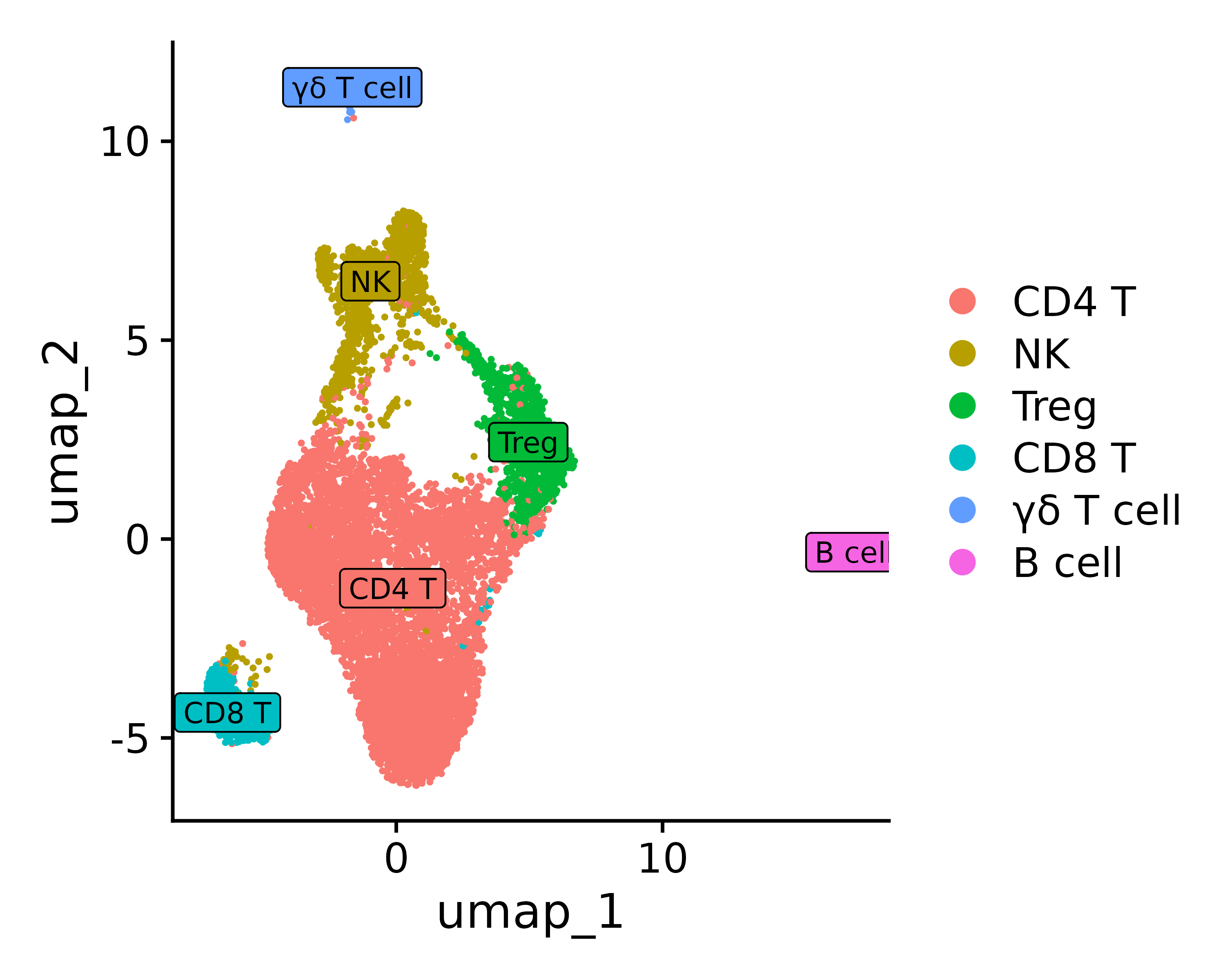
p3 = DimPlot(seurat.final, reduction = "umap", label = TRUE, label.size = 3, pt.size = .3, label.box = T)
ggsave("annotation_umap.png", p, width = 5, height =4 , dpi=600)
ggsave("annotation_umap2.png", p3, width = 5, height =4 , dpi=600)