Command Palette
Search for a command to run...
構造・配列・機能の関係性に基づくタンパク質言語モデルの分類の再定義:李明塵博士がタンパク質言語モデルを詳細に解説

上海交通大学の第3回「AIバイオエンジニアリングサマースクール」が2025年8月8日から10日まで正式に開校しました。このサマースクールには、人工知能(AI)とバイオエンジニアリングの統合開発に焦点を当て、世界中の70以上の大学、10以上の科学研究機関、10以上の業界をリードする企業から200人を超える若い才能、科学研究者、業界代表者が集まりました。
その中で、「AIアルゴリズムの最前線」の授業部門では、上海交通大学自然科学研究所の洪亮研究グループの博士研究員である李明塵氏が、「タンパク質とゲノムの基本モデル」をテーマに、機能予測、配列生成、構造予測などにおけるタンパク質言語モデルの最先端の成果、および拡張法則とゲノムモデルに関する関連研究の進歩を皆と共有しました。

HyperAIは、李明塵博士の素晴らしい講演を、本来の趣旨を損なうことなく編集・要約しました。以下は講演のハイライトです。
タンパク質言語モデルの新しい分類:タンパク質の構造、配列、機能の関係
タンパク質は化学工学、農業、食品、化粧品、医薬品、検査など幅広い分野で応用されており、その市場価値は数兆ドルを超えています。簡単に言えば、タンパク質言語モデリングは確率分布の問題です。これは、アミノ酸配列が自然界に存在する確率を決定し、それに応じてサンプリングすることと同等です。膨大なデータによる事前学習により、モデルは自然界に見られる確率分布を効果的に表現できるようになります。
タンパク質言語モデルには 3 つのコア機能があります。
* タンパク質配列を高次元ベクトルとして表現する学習プロセス
* アミノ酸配列の合理性を決定する
* 新しいタンパク質配列を生成する
多くの研究論文では、タンパク質言語モデルをTransformerアーキテクチャに基づいて分類し、Transformer EncoderベースとTransformer Decoderベースのいずれかに直接分類しています。この分類は生物学研究者にとって理解しにくく、しばしば混乱を招きます。そこで、新しい分類方法を紹介します。タンパク質の構造、配列、機能の関係に基づく分類。
タンパク質の配列とは、アミノ酸配列のことです。アミノ酸配列が分かれば、研究室や工場で合成し、実用化することができます。タンパク質の構造も同様に重要です。タンパク質の機能は、三次元空間における特定の構造に由来し、それが微視的レベルでの機能を可能にしています。
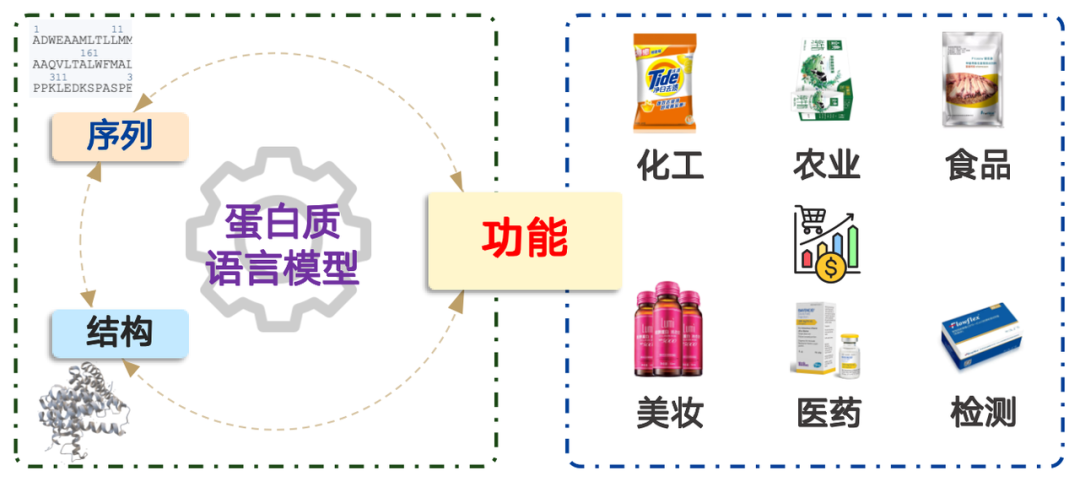
この考えに基づいて、タンパク質言語モデルは次の 4 つのカテゴリに分類できます。
1. シーケンス → 関数:特定のアミノ酸配列の機能を予測する、すなわち、 機能予測モデル。
2. 関数 → シーケンス:与えられた機能に応じて対応するアミノ酸配列を設計する。 生成モデルそして マイニングモデル。
3. シーケンス → 構造:アミノ酸配列に基づいて構造を予測することを、通常 「構造予測モデル」ノーベル賞を受賞したAlphaFoldもこのタイプのモデルに属します。
4. 構造 → シーケンス:与えられたタンパク質構造に基づいて対応する配列を設計することを、通常 「逆折りモデル」。
応用シナリオと技術パス:4つの主流モデルの分析
「シーケンス→関数」
「シーケンス→関数」を理解する最も簡単な方法は、教師あり学習です。
まず、最も基本的な機能予測モデルは、タンパク質配列をベクトルとして表現し、特定のデータセットで学習させることです。例えば、タンパク質の融点を予測したい場合、まず大量のタンパク質融点ラベルを収集し、学習セット内のすべてのタンパク質配列を高次元ベクトルに変換し、教師あり学習法を用いて学習させる必要があります。最後に、テストセットまたは予測セット内の配列に対して推論を実行し、機能を予測します。このアプローチは幅広いタスクに対応でき、現在注目されている研究テーマであり、比較的簡単に結果を得ることができます。
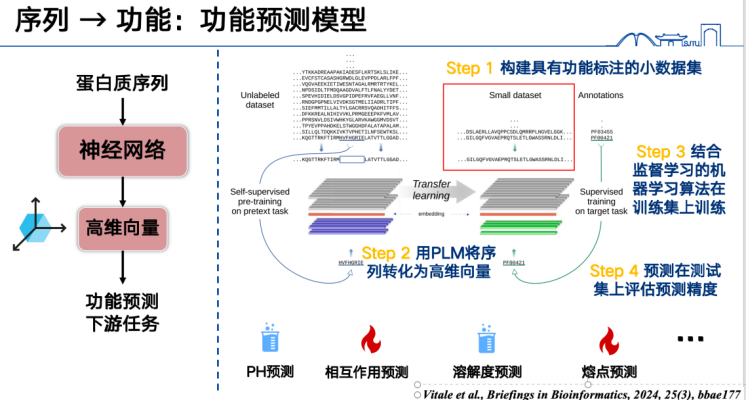
第二に、タンパク質言語モデルは変異機能を予測することもできます。基本的な考え方は、タンパク質配列内の特定のアミノ酸に何らかの変更を加え、タンパク質言語モデルを使用してその変更が「合理的」かどうかを判断することです。
ここでの「合理的」とは、現実世界における論理的な適合性ではなく、アミノ酸の変化が天然のタンパク質配列の確率分布に合致するかどうかを指します。この確率分布は、多数の実際のアミノ酸配列統計から得られ、これらのアミノ酸分布自体が数千万年にわたる進化の産物です。
タンパク質言語モデルは、訓練中にこれらの進化法則を学習し、変異がこれらの法則に従うか逸脱するかを判断できます。数学的には、この判断は変異前後の2つの配列の確率の比に変換できます。計算を容易にするため、この比はしばしば対数化され、減算形式に変換されます。
言語モデルで使用される変異体と野生型タンパク質間の尤度比は、変異の影響の強さを推定することができます。この考え方は、2018年にNature Methods誌に掲載されたDeepSequenceモデルを紹介する論文で初めて実証されましたが、当時のモデルは比較的小規模でした。その後、2021年にESM-1vモデルによって、タンパク質言語モデルも尤度比を用いて変異の影響を効果的に予測できることがさらに実証されました。
タンパク質変異機能予測モデルの精度を評価するには、ベンチマークが必要です。
ベンチマークとは、精度を測定するために収集される小規模なデータセットです。例えば、ハーバード大学医学部とオックスフォード大学が共同開発したProteinGymは、最も広く使用されているベンチマークです。このベンチマークには、217種類の変異タンパク質と数百万の変異配列のデータが含まれています。研究者は、タンパク質言語モデルを用いてこれらの変異配列それぞれにスコアを割り当て、モデルの予測スコアと実際のスコアを比較します。相関が高いほど、モデルの性能が優れていることを示します。
ただし、ProteinGym は、スループットが高く、精度が低いベンチマークです。実験条件によって制限はあるものの、大規模にテストすることは可能ですが、精度には限界があります。実験を繰り返すと、結果と元のデータとの相関関係に誤差が生じ、評価結果が実際のアプリケーションにおけるモデルの性能を正確に反映しなくなる可能性があります。
この問題を解決するには、私たちは、VenusMutHub のような低スループット、高精度の小規模サンプル ベンチマークを開発しました。データ量は多くありませんが、各データは比較的正確であり、繰り返し実験した結果はほぼ一貫しており、実際のアプリケーションシナリオに近いものとなっています。
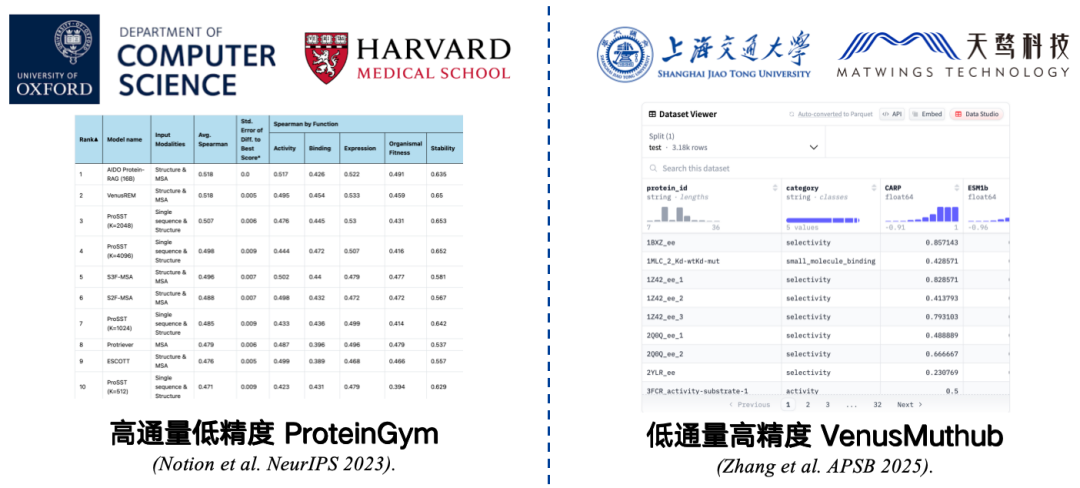
* 紙のアドレス:Zhang L, Pang H, Zhang C, et al. VenusMutHub: 小規模実験データに基づくタンパク質変異効果予測因子の体系的評価[J]. Acta Pharmaceutica Sinica B, 2025, 15(5): 2454-2467.
さらに、構造を導入することで、タンパク質言語モデルの変異予測精度を向上させることができます。昨年、私たちのチームはNeurIPSでタンパク質言語モデルであるProSSTモデルに関する論文を発表しました。このモデルは、アミノ酸配列と構造化配列の両方を用いて、マルチモーダル事前学習を行います。ProSSTは、最大規模のゼロショット変異予測ベンチマークであるProteinGymベンチマークで1位を獲得しました。
* 紙のアドレス:Li M, Tan Y, Ma X, et al. ProSST: 量子化構造と分離アテンションを用いたタンパク質言語モデリング[C]. Advances in Neural Information Processing Systems, 2024, 37: 35700-35726.
実験や設計を行う際には、「どのモデルを使用すればよいのか?」や「ユーザーとしてどのように選択すればよいのか?」といった疑問に直面することがよくあります。
今年発表された研究では、私たちのチームは、ターゲット配列に対するタンパク質言語モデルの複雑さが、変異予測タスクにおけるその精度を大まかに反映できることを発見しました。この手法の利点は、標的タンパク質の変異データを必要とせずに性能推定値を提供できることです。具体的には、パープレキシティが低いほど、モデルは配列をより深く理解しており、多くの場合、その配列に対する変異予測の精度が向上します。
この考えに基づき、私たちはアンサンブルモデルVenusEEMを開発しました。このモデルは、パープレキシティに基づいてモデルに重み付けするか、パープレキシティが最も低いモデルを直接選択します。これにより、変異予測の精度が大幅に向上します。どちらの戦略を採用しても、最終的な予測スコアは比較的安定しており、誤ったモデル選択による大幅な性能低下を防ぎます。
* 紙のアドレス:Yu Y, Jiang F, Zhong B, et al. エントロピー駆動型ゼロショット深層学習によるウイルスタンパク質のモデル選択[J]. Physical Review Research, 2025, 7(1): 013229.
最後に、「配列から機能へ」という研究方向において、前述のモデルに加え、昨年、私たちのチームは新たな反復型高サイト変異設計モデル「PRIME」を開発しました。具体的には、まず9,800万個のタンパク質配列を用いて大規模タンパク質言語モデルを事前学習しました。高サイト変異予測タスクでは、まず低サイト変異データを取得し、それをタンパク質言語モデルに入力して関数ベクトルにエンコードしました。次に、この関数ベクトルに基づいて回帰モデルを学習し、高サイト変異を予測しました。この反復反応により、わずか 2 ~ 3 回の実験で優れたタンパク質製品を開発できます。
* 紙の住所:Jiang F, Li M, Dong J, et al. 安定性と活性を高めたタンパク質を設計するための一般的な温度誘導言語モデル[J]. Science Advances, 2024, 10(48): eadr2641.

「関数→シーケンス」
これまで議論してきたのは、シーケンスから関数への変換です。では、逆に関数からシーケンスを演繹できるかどうか考えてみましょう。
シーケンスと関数の間には、順問題と逆問題があります。順問題は明確な答えを見つけることですが、逆問題は広大な実行可能空間内で解ける解を探すことです。関数からシーケンスを生成することは、まさにこの逆問題です。その理由は、シーケンスは通常、1つまたは少数の関数にしか対応しないのに対し、1つの関数は全く異なる様々なシーケンスによって実装できるためです。さらに、逆問題には信頼できるベンチマークが存在しません。モデルが特定の関数からシーケンスを生成する場合、その精度は通常、実験的にしか検証できません。

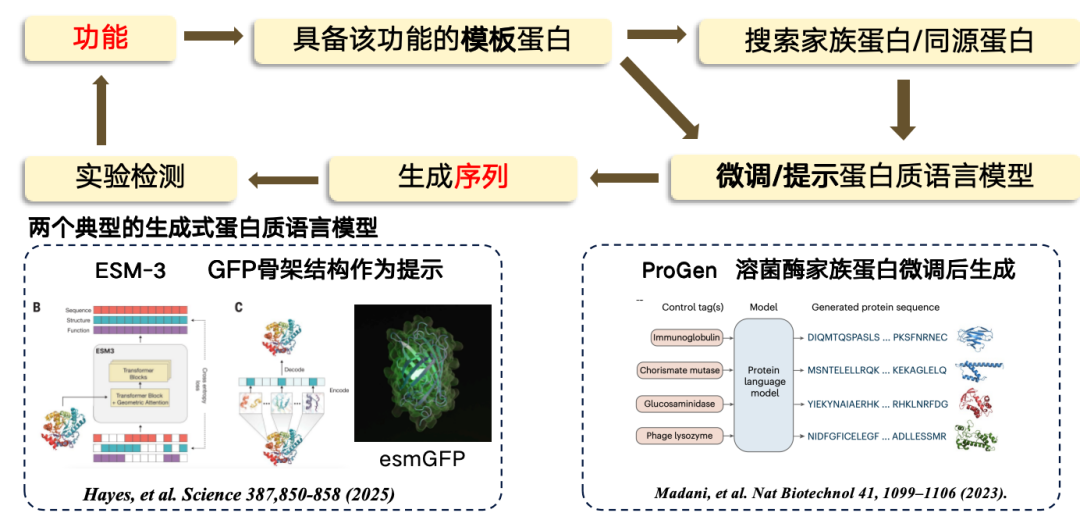
本研究では、機能から配列までの設計は、主にテンプレートベースのアプローチを採用します。テンプレートタンパク質が特定の機能を持つことが分かっている場合、それを基に新しい領域を発見または生成することができます。プロセスは、まず機能からテンプレート配列に進み、次にテンプレートタンパク質からいくつかのファミリータンパク質/相同タンパク質を検索し、次にタンパク質言語モデルを微調整し、微調整された言語モデルを使用して新しい配列領域を生成し、最後に実験テストを実施するというものです。
現在、最も代表的な 2 つの生成タンパク質言語モデルは次のとおりです。
*ESM-3は緑色蛍光タンパク質(GFP)をテンプレートとして使用して生成されますが、生成されるタンパク質の機能性は低くなります。
* ProGenは、ChatGPTに類似した、機能的手がかりに基づいて生成可能な純粋自己回帰言語モデルです。リゾチームのタンパク質構造を微調整することで生成されます。
新しいタンパク質配列を直接生成するだけでなく、膨大な量の既存のタンパク質配列から直接検索することもできます。テンプレートタンパク質は高次元空間にエンコードされ、ベクトル間の距離によって2つのタンパク質が同じ機能を持つかどうかが判断されます。最終的に、データベースから結果が取得されます。このアプローチの原理は、高次元空間における2つのタンパク質のエンコードまたはベクトル間の距離が、2つのタンパク質が類似した機能を持つかどうかを大まかに反映できるというものです。
下の図は、タンパク質言語モデルマイニングの典型的な例を2つ示しています。1つ目はウェストレイク大学が開発したESM-Ezyで、ESM-1bモデルを用いてベクトル検索を行い、複数の表現をマイニングしてインフィリングを行います。2つ目は、高効率PET加水分解酵素をマイニングする大規模モデルVenusMineです。
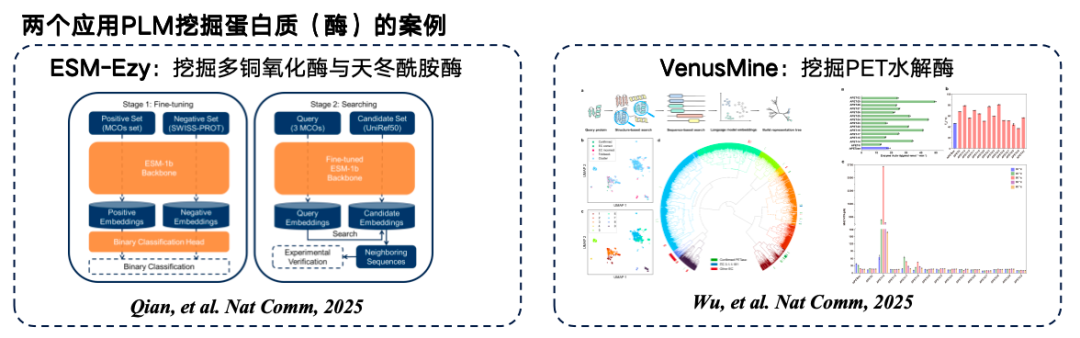
* 紙の住所:Wu B, Zhong B, Zheng L, et al. タンパク質言語モデルを活用した構造ベースの高効率かつ堅牢なPET加水分解酵素の発見[J]. Nature Communications, 2025, 16(1): 6211.
「関数 → シーケンス」に加えて、関数とシーケンスの間に「メディエーター」を追加することもできます。
* 構造が仲介として使用される場合: タンパク質構造は機能に基づいて推論され (RFdiffusion などの一般的なツール)、生成された構造は逆タンパク質フォールディング言語モデル (ProteinMPNN など) に入力され、最終的にシーケンスが生成されます。
* 自然言語を媒体として用いる場合:例えば、研究論文「テキスト誘導型タンパク質設計フレームワーク」で説明されている手法では、比較学習を通じて自然言語とタンパク質配列を高レベル空間にアラインメントします。そして、この高レベル空間において、自然言語のガイダンスを用いて直接タンパク質配列を生成することができます。
シーケンス → 構造
配列から構造への方向において、最も古典的なモデルは間違いなくAlphaFoldです。では、なぜ構造予測にタンパク質言語モデルが依然として必要なのでしょうか?主な理由は、速いことです。
AlphaFold の速度低下の主な原因は、MSA(多重配列アライメント)検索において、大規模なデータベースの検索にCPUを依存していることです。GPUアクセラレーションは可能ですが、実際のアクセラレーションはさらに遅くなります。また、AlphaFold はフォールディングプロセス中にテンプレートマッチングも必要とし、これもかなりの時間を消費します。これらの2つのモジュールをタンパク質言語モデルに置き換えることで、構造予測プロセスを大幅に高速化できます。しかしながら、現在発表されている研究によると、タンパク質言語モデルに基づく構造予測の精度は、ほとんどの評価指標において、AlphaFold モデルよりも依然として低いことが示されています。
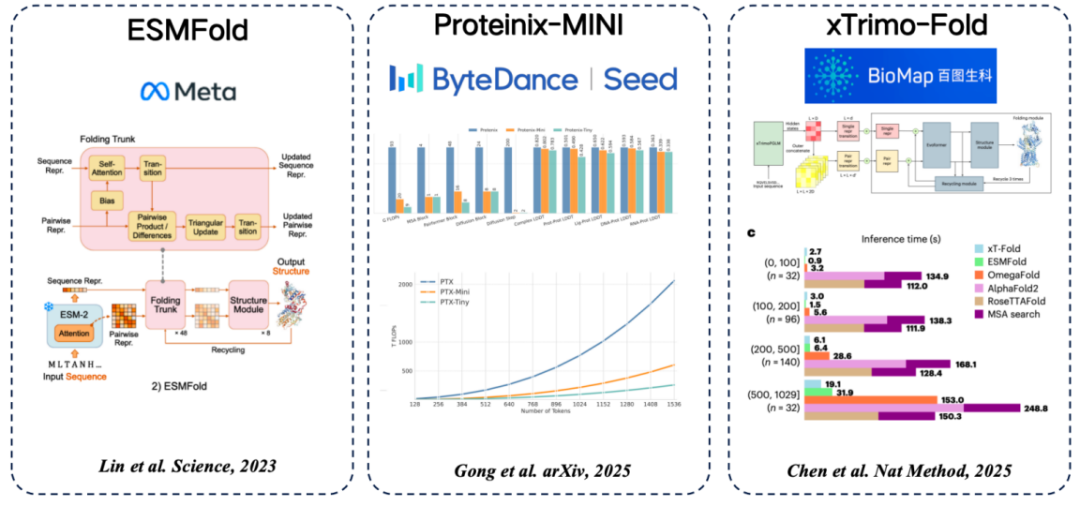
配列から構造まで、一般的なタンパク質言語モデルがいくつかあります。MSAの代わりにタンパク質言語モデルから抽出した特徴を使用するという一般的なアイデアが採用されています。
* ESMFold(Meta):タンパク質言語モデルを用いてタンパク質構造を直接予測する初の手法であり、MSA検索に依存せずに高い精度を実現。
* Proteinix-MINI (ByteDance): MSA の代わりにタンパク質言語モデルを使用します。これも非常に高速な結果を実現し、AlphaFold 3 モデルに近い予測精度を備えています。
* xTrimo-Fold (Baidu Biosciences): MSA の代わりに 1000 億パラメータ モデルの機能を使用することで、検索を高速化します。
構造 → シーケンス
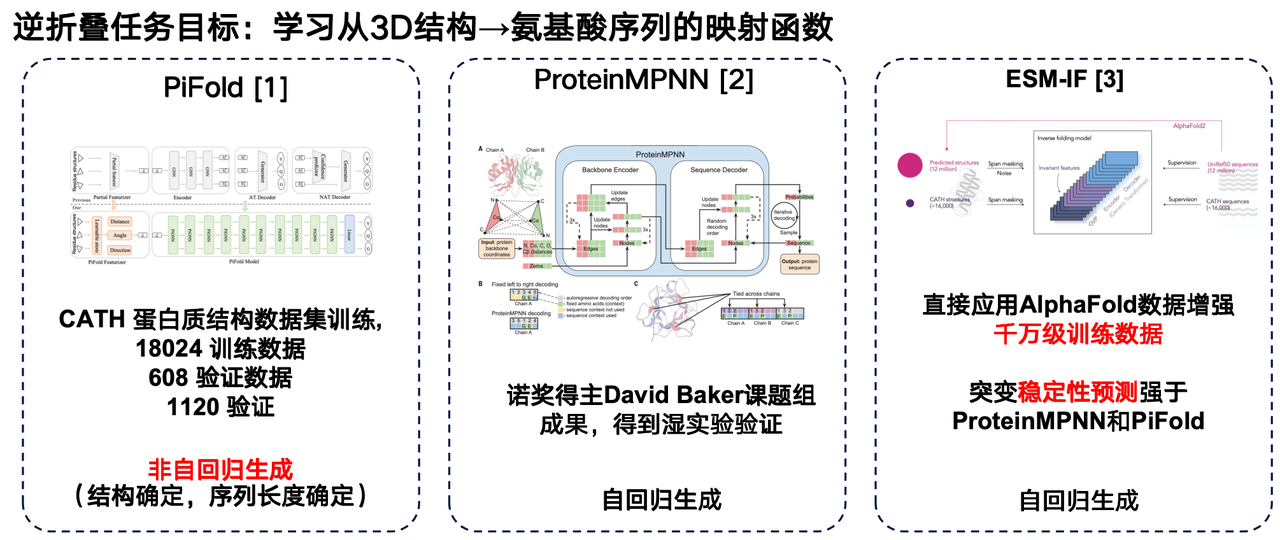
構造は既知の機能に基づいて設計されていますが、それを実験室でどのように合成するのでしょうか?さらに、これをアミノ酸の配列に変換する必要があります。これが、前述の「逆折り畳み言語モデル」です。
逆フォールディング言語モデルは、AlphaFoldの「逆問題」と考えることができます。アミノ酸配列から3D構造を予測するAlphaFoldとは異なり、逆フォールディングモデルは、タンパク質の3D構造からアミノ酸配列へのマッピング関数を学習することを目的としています。
この分野におけるいくつかの研究成果をご紹介したいと思います。まず1つ目は、ウェストレイク大学の研究チームによるPiFoldモデルです。このアーキテクチャにおける大きな革新は、非自己回帰生成法の採用です。
2つ目は、David Bakerの研究グループが開発したProteinMPNNです。最も広く使用されている逆フォールディングモデルの一つであり、自己回帰生成を用いて個々のタンパク質構造をグラフニューラルネットワークでエンコードし、アミノ酸配列を一つずつ生成します。
MetaのESM-IFもまた、重要な進歩です。その最大の特徴は、AlphaFoldによって予測された膨大な構造データを活用し、数千万のタンパク質配列に対応する3次元構造を均一に予測することで、極めて大規模なトレーニングセットを構築したことです。ESM-IFのトレーニングデータは数千万ユニットに達し、モデルのパラメータ数は1億を超えています。これにより、このモデルは逆フォールディングタスクを実行するだけでなく、変異の安定性予測においても優れた性能を発揮します。
タンパク質言語モデルを強化するための複数のアプローチ
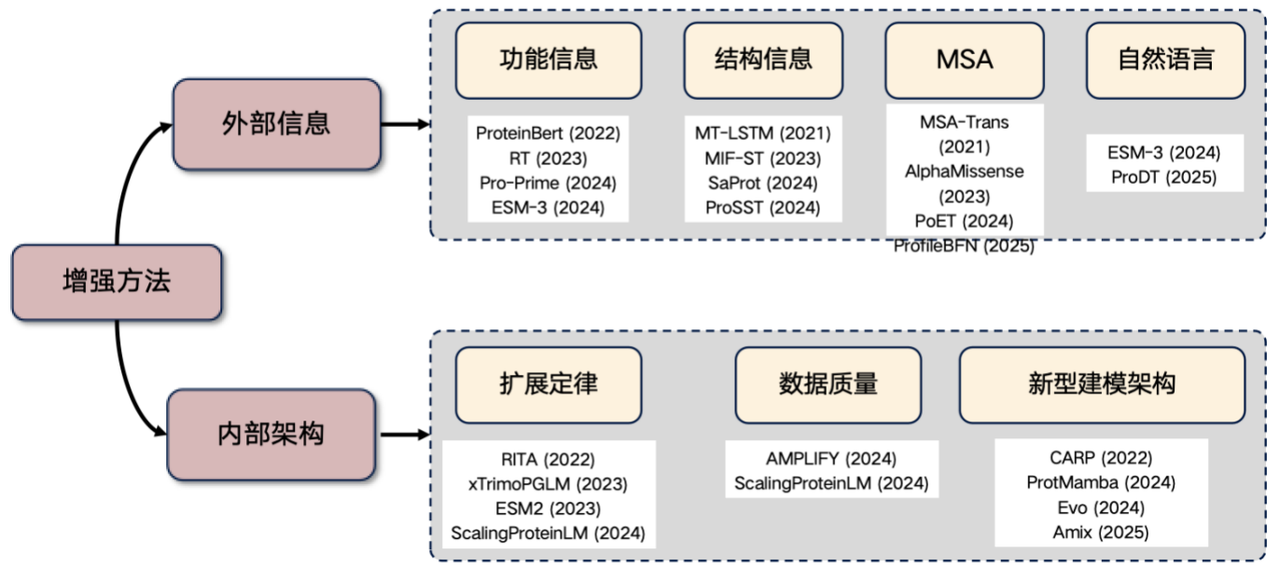
最後に、現在非常に注目されている研究分野である、タンパク質言語モデルの拡張について触れておきます。この分野で研究を行う予定であれば、以下のアイデアから始めることができます。外部情報を導入し、内部構造を改善します。
1. 外部情報の紹介
* 機能情報:例えば、温度やpHなどの特徴量をTransformerに入力します。これらの情報は、モデル入力に明示的に組み込むことも、学習されたアクションを通じて組み込むこともでき、タンパク質言語モデルの性能を向上させることができます。
* 構造情報: 3次元構造や構造化された配列情報を紹介します。
* MSA情報:多重配列アライメント(MSA)は非常に有用な情報です。これを言語モデルに導入することで、パフォーマンスが大幅に向上することがよくあります。
* 自然言語情報:近年、自然言語情報を取り入れようとする研究も行われていますが、この方向性はまだ模索中です。
2. 内部アーキテクチャの改善
* スケーリング法則: モデルパラメータの数とトレーニングデータのサイズを大幅に増やすことで、パフォーマンスが向上します。
* データ品質の向上: データのノイズを削減し、精度を向上します。
* 新しいアーキテクチャの探索: CARP、ProtMamba、Evo アーキテクチャなど。
近年、タンパク質構造情報を利用してモデルのパフォーマンスを向上させることが、注目されている研究分野となっています。
最も初期の代表的な研究の一つは、2021年の論文「タンパク質言語の学習:進化、構造、機能」であり、構造情報を用いてタンパク質言語モデルの機能を強化する方法を示しました。その後、SaProtモデルは巧妙なアプローチを提案しました。タンパク質のアミノ酸語彙と、Foldseekによってタンパク質構造用に生成された20個の仮想構造語彙を連結し、最終的に400語(20×20)の複合語彙を生成します。この語彙はマスク付き言語モデルの学習に使用され、優れた精度を達成しました。
当チームは、タンパク質の配列と構造を対象とするマルチモーダル事前学習モデルProSSTを独自に学習しました。このモデルは、タンパク質の連続構造を離散トークン(2,048種類のトークン)に変換することで、構造情報の離散表現を実現します。
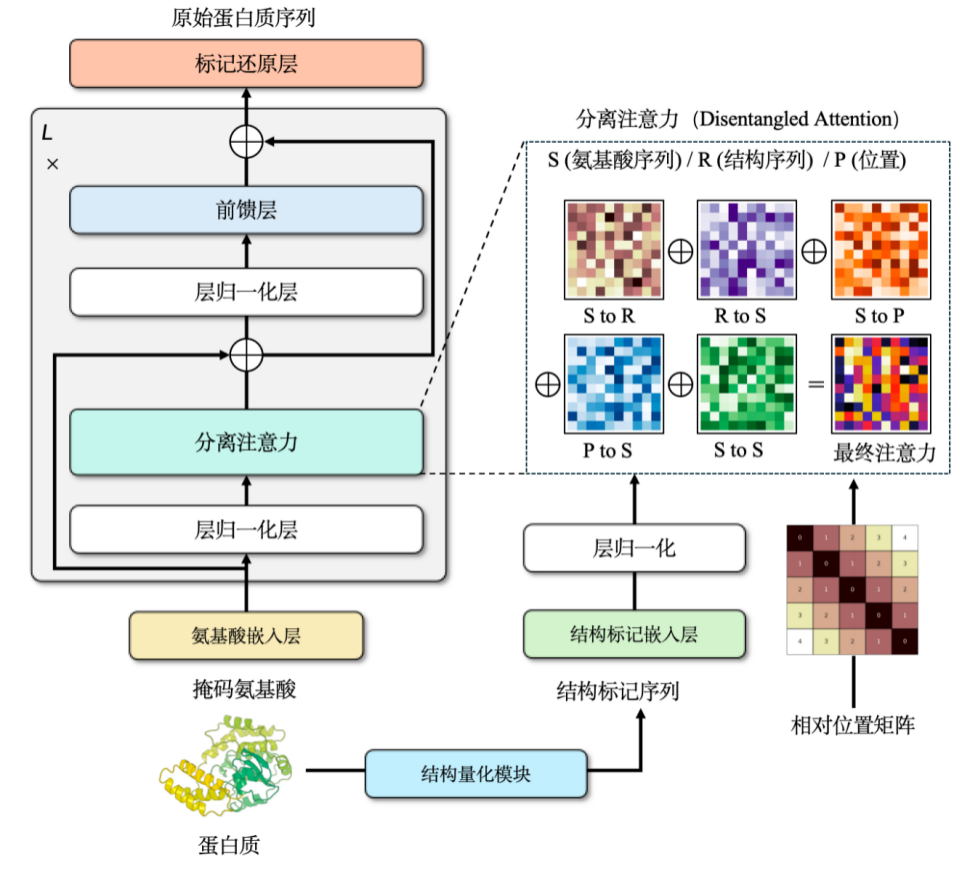
タンパク質言語モデルに構造情報を組み込むことで、モデルのパフォーマンスを大幅に向上させることができます。しかし、このプロセス中に問題が発生する可能性があります。AlphaFoldによって予測された構造データを直接学習に用いると、学習セットの損失は徐々に減少する一方で、検証セットまたはテストセットの損失は徐々に増加するという問題です。この問題を解決する鍵は、構造情報を正規化することです。簡単に言えば、複雑なデータを簡素化して、モデル処理に適したものにすることを意味します。
タンパク質構造は通常、3次元空間における連続座標として表現されます。これを離散的な整数列に変換することで簡略化する必要があります。この目的のため、グラフニューラルネットワークアーキテクチャを用い、ノイズ除去エンコーダを用いて学習させることで、最終的に約2,048トークンからなる離散構造語彙を構築しました。
構造と配列の情報により、私たちは、この 2 つを組み合わせるためにクロス アテンション メカニズムを選択しました。これにより、修正されたTransformerモデルはアミノ酸配列と構造配列の両方を入力できるようになります。事前学習段階では、このモデルを言語モデル開発タスクとして設計しました。トレーニング データには、パラメーター サイズが約 1 億 1,000 万である 1,880 万を超える高品質なタンパク質構造が含まれています。このモデルは当時最先端の結果を達成し、その後新しいモデルに追い抜かれたものの、発表時点では同クラスとしては依然として最高の結果を保持していました。
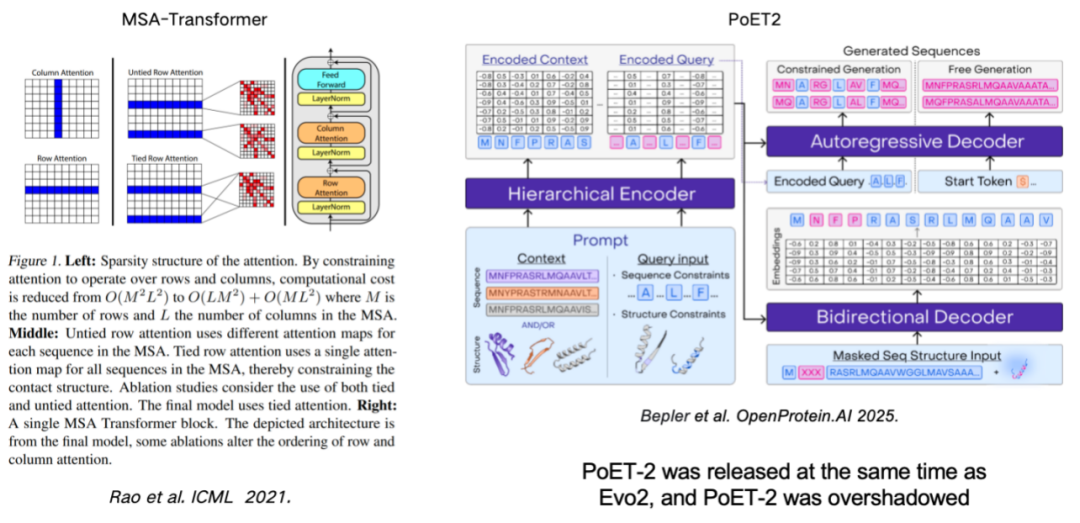
MSA (多重配列アライメント) を使用してタンパク質言語モデルを強化することも、モデルのパフォーマンスを向上させる重要な手段です。この研究は、行と列のルールを導入することでMSA情報をモデルに効果的に組み込んだMSA-Transformerに遡ります。最近リリースされたPoET2モデルは、階層型エンコーダを用いてMSA情報を処理し、それをフルパスモデルアーキテクチャに統合しています。大規模学習により、優れた性能を示しました。
スケーリングの法則: より大きなモデルは常により強力ですか?
いわゆるスケーリング則は自然言語処理の分野から生まれたもので、普遍的な法則を明らかにしています。モデルのパフォーマンスは、パラメータスケール、トレーニングデータ量、コンピューティングリソースの増加に伴って向上し続けます。
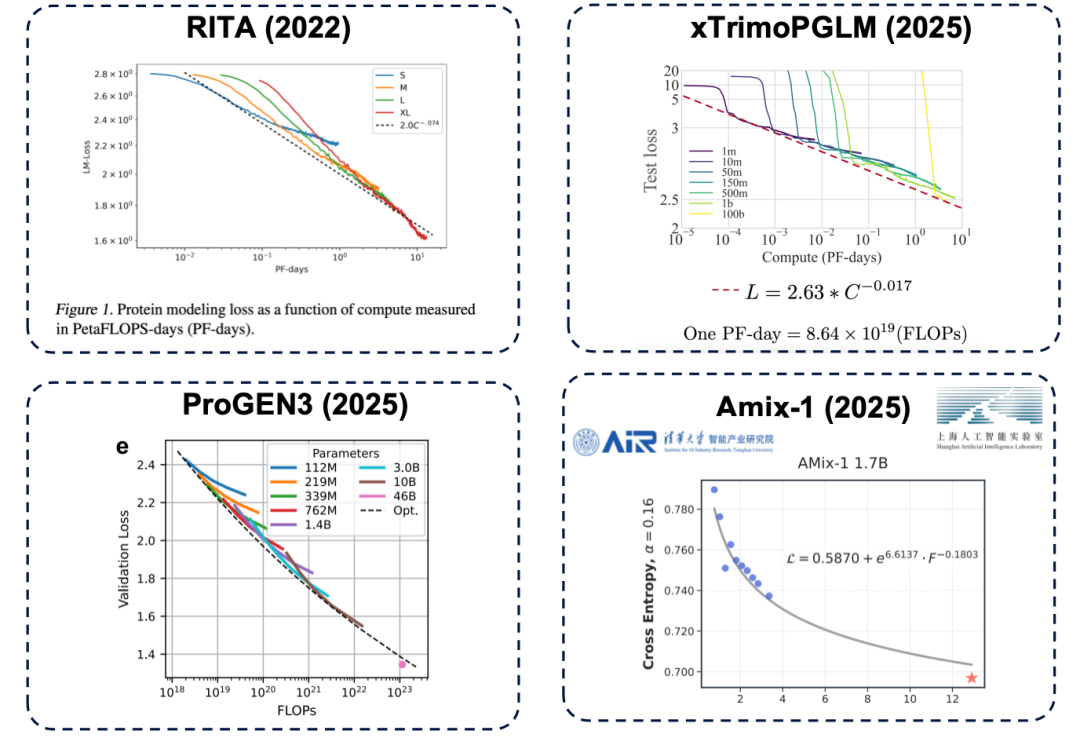
パラメータサイズは、モデル性能の上限を決定する重要な要素です。パラメータ数が不足している場合、たとえ計算リソース(平たく言えば「より多くの費用」)を投入しても、モデル性能はボトルネックに達してしまいます。この原理はタンパク質言語モデルの分野でも存在し、RITA、xTrimoPGLM、ProGEN3、Amix-1といった代表的な研究を含む数多くの研究によって確認されています。
* RITA モデル: オックスフォード大学、ハーバード大学医学部、LightOn AI によって開発されました。
* xTrimoPGLM モデル: Baitu Bioscience チームによって開発され、モデル パラメータを約 1000 億に拡張します。
* ProGEN3 モデル: Profluent Biotech チームによって開発されました。
* Amix-1 モデル:清華大学知能産業研究所と上海人工知能研究所が提案した、ベイズフローマッチングネットワークアーキテクチャを採用し、拡張法則も備えています。
先ほど述べた「スケーリング則」は、事前学習プロセスを指します。しかし、タンパク質研究では、最終的には下流のタスクのパフォーマンスに焦点が当てられます。このことから、次のような疑問が生じます。事前トレーニングのパフォーマンスの向上は、必ず下流のタスクに役立ちますか?
xTrimoPGLM 評価では、研究チームは約 44% のダウンストリーム タスクにおいて、「事前トレーニングのパフォーマンスの向上とダウンストリーム パフォーマンスの強化」の間には確かに正の相関関係があることを発見しました。
同時に、Amix-1モデルは構造予測タスクにおいて創発的な能力を発揮しました。これは、小規模なモデルでは問題を全く解くことができないものの、モデルのパラメータサイズが一定の臨界点を超えるとパフォーマンスが急激に向上するタスクを指します。今回の実験では、この現象は特に構造予測タスクにおいて顕著で、パラメータサイズが臨界点を超えるとパフォーマンスの向上が「崖のような赤い線」を示しました。
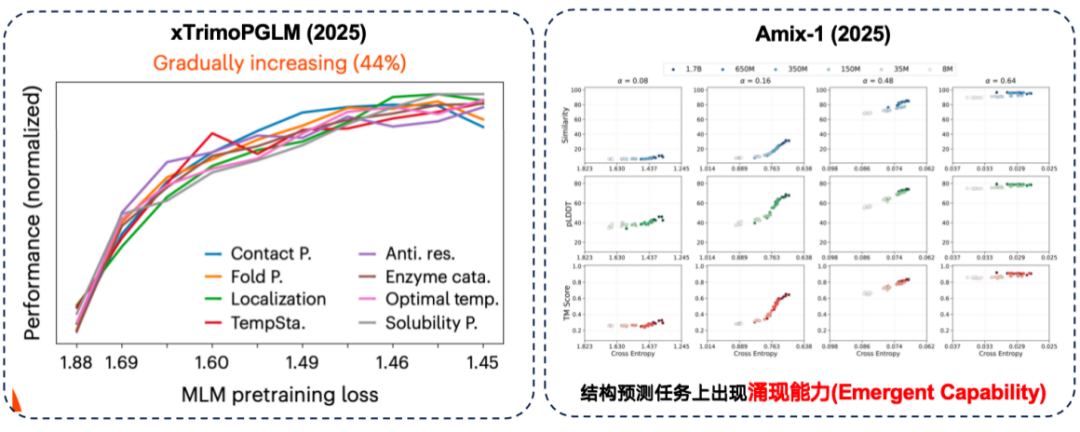
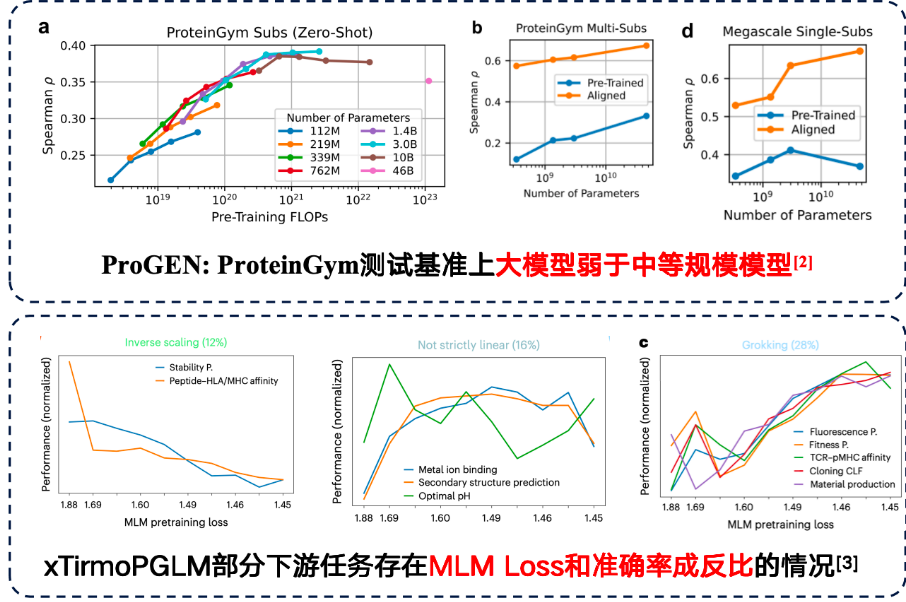
ただし、一部のタスクでは、大規模なモデルによって下流のパフォーマンスが向上する場合があります。しかし、下流のタスクでは逆スケーリング則も発見されました。つまり、モデルが小さくなるほど、パフォーマンスは向上します。
研究によると、トレーニングデータ自体にノイズが多い場合、モデルのパラメータ数を単純に増やしても結果が改善されないことが示されています。そのため、データ品質にはより一層の注意を払う必要があります。ProteinGymベンチマークのタンパク質変異予測タスクでは、中規模モデルの方が精度の点で優れたパフォーマンスを示しました。さらに、xTirmoPGLMの開発チームは、トレーニング前のパフォーマンスと下流タスクのパフォーマンスが一致しない、非正相関のケースもいくつか発見しました。
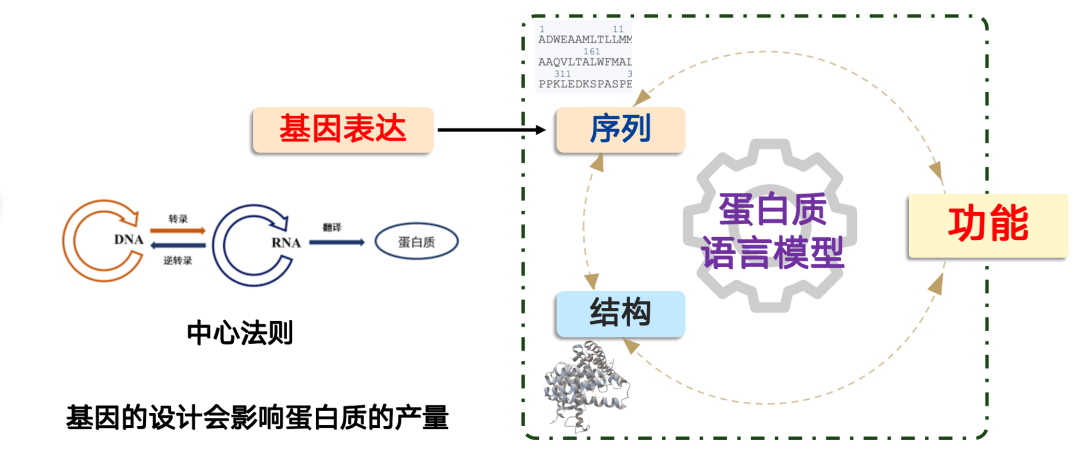
ゲノムモデリング:DNA設計からタンパク質収量最適化まで
ゲノムモデルが解決する問題は、「タンパク質をどうやって生成するか」です。
合成生物学では、タンパク質の生成は、分子生物学のセントラルドグマ「DNA → RNA → タンパク質」に従います。細胞内では、このプロセスは細胞体によって制御されており、遺伝子を設計することでこのプロセスを完了することができます。しかし重要なのは、遺伝子設計がタンパク質の生産に直接影響を与えるということです。
実用化においては、タンパク質が優れた機能性能を有しているにもかかわらず、遺伝子設計の不備により発現レベルが極めて低く、産業化や大規模応用のニーズを満たせないという状況にしばしば遭遇します。このような場合、AIモデルが役割を果たします。
AIモデルの使命は、タンパク質配列から直接DNA配列を設計する方法を推測し、その生産性を向上させることです。私たちのチームが提案するモデルProDMMは、事前学習戦略に基づいており、以下の2つのフェーズで構成されています。
第一段階では、タンパク質とDNAの表現を学習するために、ジョイント事前学習が用いられます。入力にはタンパク質配列とDNA配列が含まれ、Transformerアーキテクチャを用いて言語モデルが学習されます。目標は、タンパク質、コドン、DNA配列の表現を同時に学習することです。第二段階では、タンパク質からコード配列(CDS)への変換といった下流タスクを用いて生成タスクを学習します。タンパク質が与えられれば、DNA配列を生成できます。
* 紙のアドレス:Li M, Ren Y, Ye P, et al. 統合マルチモーダルシーケンスモデリングを用いたタンパク質-DNA相互依存性の解明[J]. bioRxiv, 2025: 2025.02. 26.640480.
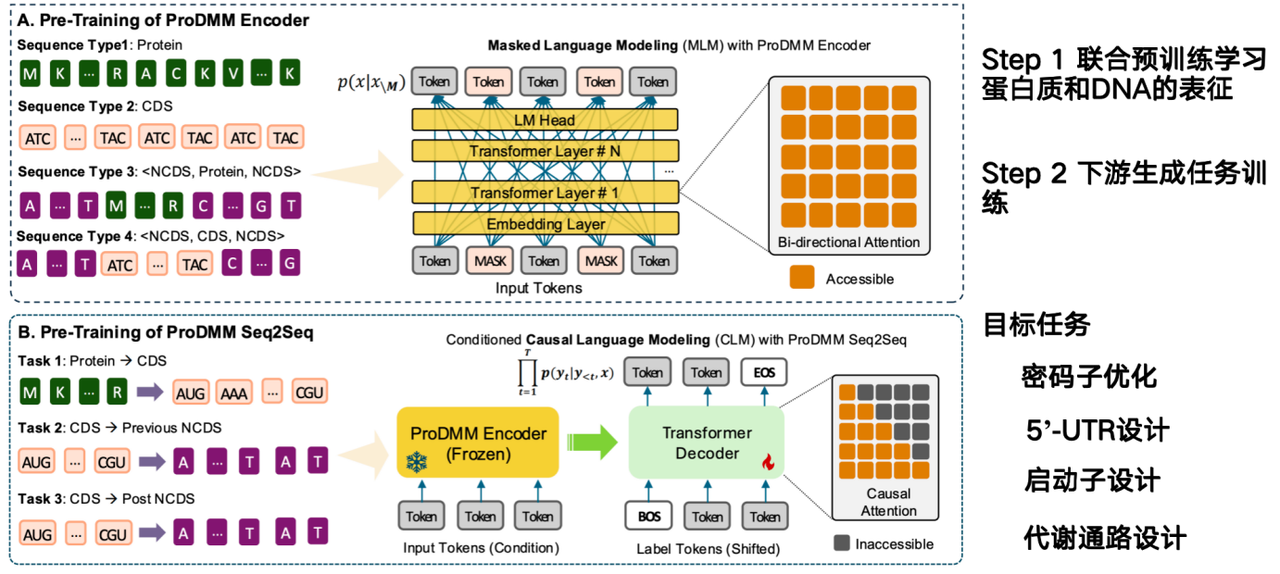
コドンから非コード DNA (NCDS) までのプロジェクトの目標は、コドンの最適化、5'-UTR 設計、プロモーター設計、および代謝経路設計を完了することです。
代謝経路の設計は、遺伝子内の複数のタンパク質が協調して特定の産物を合成することを伴います。代謝経路全体の産物を最適化する必要があり、これはゲノムモデルに特に適した作業です。タンパク質モデルは単一のタンパク質のみを最適化し、コンテキストに依存しないからです。しかし、ゲノムモデルが直面する大きな課題は、細胞環境における相互関係を考慮する必要があることです。これが現在、ゲノムモデルにとって最大の課題となっています。
リー・ミンチェン博士について
この共有セッションのゲストスピーカーは、上海交通大学自然科学研究所のHong Liang研究グループのポスドク研究員であるLi Mingchen氏です。彼は華東科技大学でコンピュータサイエンスとテクノロジーの工学博士号と数学の理学士号を取得しています。彼の主な研究分野は、タンパク質言語モデルの事前学習とファインチューニングです。
上海市優秀卒業生、国家奨学金、そして「インターネット+」大学生イノベーション・起業コンテスト上海部門金メダルを受賞。NeurIPS、Science Advances、Journal of Cheminformatics、Physical Review Researchなどのジャーナルや会議に筆頭著者、共同筆頭著者、責任著者として計10本のSCI論文を発表し、10本のSCI論文の出版にも参加。
2023年から2024年にかけてのAI4S分野の高品質な論文と詳細な解釈記事をワンクリックで入手⬇️









