Command Palette
Search for a command to run...
スタンフォードらは、タンパク質の主鎖と側鎖の情報を同時に処理し、メッセージパッシングニューラルネットワークに基づく完全な原子構造モデリングを実現した。
タンパク質側鎖構造とは、タンパク質中のアミノ酸残基の側鎖が三次元空間においてどのように特定の空間配置をとるかを指します。タンパク質側鎖構造を研究することは、タンパク質の構造と機能の関係を理解するのに役立ち、タンパク質工学、医薬品設計などの分野において大きな応用価値を有します。しかしながら、現在のディープラーニングに基づくタンパク質配列設計手法は、主に固定された主鎖タンパク質配列設計に焦点を当てており、その多くは配列生成時にタンパク質側鎖構造をモデル化することができません。主要な側鎖の相互作用は、タンパク質におけるタンパク質側鎖の立体配座の役割を無視して、主鎖の形状と既知のアミノ酸配列タグのみに基づいて推測されます。
このギャップを埋めるために、スタンフォード大学とカリフォルニア州パロアルトのアーク研究所のチームが、私たちは共同で、各アミノ酸残基の配列同一性と側鎖構造を明示的にモデル化できる新しいタンパク質配列設計法、FAMPNN (Full-Atom MPNN) を提案しました。このモデルは、グラフニューラルネットワーク(GNN)に基づくメッセージパッシングアーキテクチャを採用し、改良されたMPNN(メッセージパッシングニューラルネットワーク)およびGVP(ジオメトリックベクトルパーセプトロン)モジュールを組み合わせ、全原子エンコーディングを実現しています。また、タンパク質の主鎖と側鎖の情報を同時に処理できます。研究により、FAMPNNは全原子構造を明示的にモデル化することで、タンパク質配列設計の品質と実験予測の精度を大幅に向上させることが示されています。
「FAMPNN による全原子タンパク質配列設計のための側鎖コンディショニングとモデリング」と題された研究成果が ICML 2025 に選出されました。
研究のハイライト:
* クロスエントロピーと拡散損失の目的を組み合わせて、残基の離散配列同一性と連続側鎖構造の両方のラベルごとの分布をモデル化する手法を紹介します。
* 結合分布からサンプルを生成するための軽量反復サンプリング法を実装し、全原子エンコーディングのために改良されたMPNNとGVP層を使用する。
* 研究では、FAMPNNは全原子構造を明示的にモデル化することで、配列設計と実験的タンパク質適合性予測の精度を効果的に向上できることが示されています。

用紙のアドレス:
AIフロンティアに関するその他の論文:
https://go.hyper.ai/owxf6
データセット: 多様なデータセットがモデルのトレーニングと評価を最適化します
モデルの有効性と信頼性を確保するため、研究チームはトレーニングと評価に複雑なマルチデータセットを使用しました。
この研究では主にCATH 4.2のS40データセットを使用しました。このデータセットは、タンパク質データバンク (PDB) から抽出されたドメインの厳選されたセットであり、相同性が 40% を超える冗長ドメインが削除され、トレーニング セット、検証セット、テスト セットに 8:1:1 の比率で分割されています。
PDBデータセットはPDBデータベース全体に基づいて構築されており、2021年9月30日時点で公開された構造が含まれています。研究者らは、タンパク質鎖レベルで40%の配列相同性に従ってタンパク質をクラスター化し、モデルが多重鎖タンパク質の設計を学習するためのトレーニング用に多重鎖タンパク質の例を優先順位付けしました。
CASP13、14、および 15 データセットは、主にサイドチェーンパッキングにおけるモデルのパフォーマンスを評価するために使用されます。研究チームは、MMseqs2 サジェスト検索を使用して、トレーニング データセットと検証データセットから CASP13、14、15 データセットとの相同性が 40% を超えるすべての配列を削除し、予測された側鎖と実際の側鎖間の平均二乗平均平方根偏差 (RMSD) によって側鎖パッキング パフォーマンスを測定しました。
SKEMPlv2 データセットは、タンパク質間の結合親和性に対するモデルの予測能力を評価するために使用されました。このデータセットは、数百のタンパク質間相互作用における数千の配列変異体の実験的に測定された結合親和性を照合し、処理後の最終的なデータセットは 6,649 のデータ ポイントになります。
S669、Megascale、FireProtDB データセットを使用して、タンパク質安定性に対するモデルのゼロショット予測能力を評価しました。これらのデータセットには、様々な天然タンパク質(△△G)の安定性変化の実験測定値が含まれており、安定性予測のベンチマークデータセットとして広く使用されています。メガスケールデータセットは、データセットの重複排除版です。研究チームは、トレーニングセット、検証セット、テストセットを1つのデータセットに統合し、最終的に298種類のタンパク質を含む272,712の実験データポイントを含むデータセットを取得しました。FireProtDBデータセットには、100種類のユニークなタンパク質の3,438の単一変異の自由エネルギー変化が含まれており、そのうち3,420の例が最終的に処理後に使用されました。S669データセットには、94種類のタンパク質の669の単一変異の実験測定値が含まれており、非標準アミノ酸の存在により4つの変異体がデータセットから除外されました。
CR9114、CR6261、および G6 データセットは、抗体-抗原結合親和性を予測するモデルのパフォーマンスを評価するために使用されました。このうち、CR9114データセットには16種類のアミノ酸置換のあらゆる組み合わせが含まれています。CR6261データセットには11種類のアミノ酸置換のあらゆる組み合わせが含まれており、それぞれ合計65,536個と2,048個の配列が含まれています。G6データセットには、VEGF-Aに結合するデータポイントが合計4,275個含まれています。
タンパク質の配列と側鎖構造を同時に理解できるインテリジェントツール
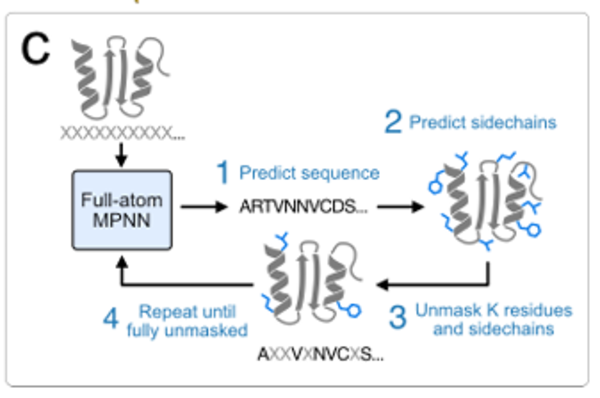
本研究の中心的な目標は、モデルがタンパク質配列と側鎖の立体構造を同時に学習できるようにすることです。この目的のために、研究チームはマスク言語モデリングを用いて、配列の一貫性に基づいてFAMPNNを学習しました。トレーニングは、カテゴリクロスエントロピー損失 (配列予測用) と拡散損失 (側鎖コンフォメーション予測用) を組み合わせて、エンドツーエンド方式で実行されます。これにより、モデルは部分的に既知の配列と側鎖座標に基づいて、マスクされた配列とそれに対応する側鎖の立体配座を同時に復元できるようになります。
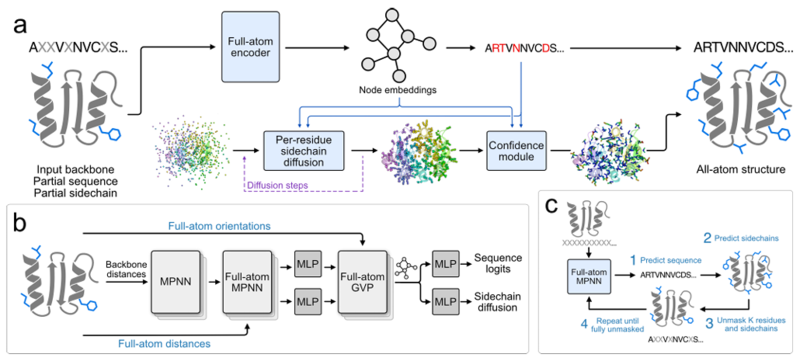
サンプリングに関しては、MaskGITに類似した反復サンプリング戦略が導入され、配列と側鎖が完全にマスクされた状態から開始し、一部の配列と側鎖トークンのマスクを段階的に予測して削除し、完全な配列と側鎖構造が得られるまで続きます。下図に示されています。
特定の設計では、側鎖座標は atom37 形式で表されます。各残基は37 x 3の固定サイズ行列で、37個の原子で構成されます。これには4つの主鎖原子(N、Cα、C、O)と33個の側鎖原子の3次元座標が含まれます。特定の原子タイプが存在しない側鎖については、ゴースト原子(残基のCα位に設定)で表されます。この手法は、アミノ酸ごとに側鎖原子の数が異なるという問題を解決します。
特徴抽出の中核として、全原子エンコーダーはエンコードにハイブリッド MPNN-GVP アーキテクチャ グラフ ニューラル ネットワークを使用します。このアーキテクチャは、不変メインチェーンエンコーダ、不変全原子エンコーダ、および等価全原子エンコーダという3つの主要コンポーネントで構成されています。最初の2つのコンポーネントは、ProteinMPNNのアーキテクチャに基づいて構築されています。不変メインチェーンエンコーダは、メインチェーン構造のみをエンコードするMPNNエンコーダと同じです。不変全原子エンコーダは、メインチェーンエンコーダMPNNエンコーダと同じMPNNデコーダーを置き換えますが、特性評価は全原子に拡張されています。最後のコンポーネントでは、改良されたGVPを使用することで、従来エンコードされていたスカラー値の原子間距離に加えて、ベクトル値の原子間配向についてもモデルが推論できるようになります。
側鎖座標生成に関しては、研究チームはトークンごとのユークリッド拡散法を採用しました。この手法の核心は、ユークリッド拡散モデル(EDM)を用いて、側鎖原子座標の連続値生成問題を解決することです。目標は、主鎖構造と周囲のアミノ酸の空間配置に一致する側鎖構造を生成することです。学習時には、まず実際の側鎖座標にランダムノイズを加え、その後、モデルはノイズレベルと既知の情報に基づいてノイズを除去し、実際の座標を復元します。推論時には、ランダムノイズ座標から始めて、モデルは徐々にノイズを除去し、実際の座標に近い側鎖座標を生成します。
同時に、タンパク質全体の回転と並進が側鎖生成に影響を及ぼさないように、トレーニング中に側鎖原子座標を主鎖原子に基づくローカル座標系に変換し、生成後にグローバル座標系に再変換します。拡散モデルの入力には、全原子エンコーダーによって抽出された特徴、予測されたシーケンス ID、および現在のノイズ レベルが含まれます。生成されたサイドチェーン座標は、下の図に示すように、モデルのトレーニングをガイドするためのジョイント損失関数でも使用されます。
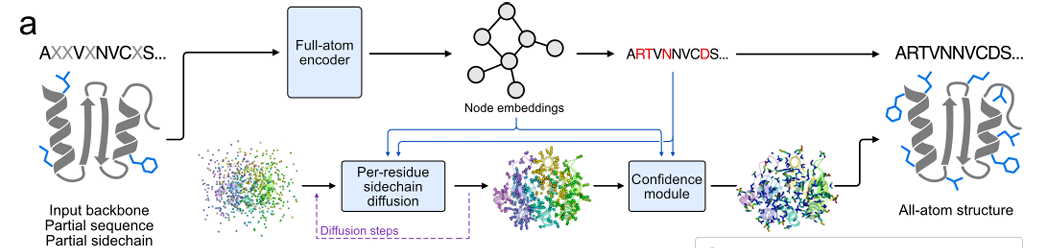
モデル予測の誤差を低減し、精度を向上させるために、研究チームはサイドチェーンパッキング誤差(Predicted Sidechain Error、pSCE)を予測するための信頼性モジュールも設計しました。具体的には、このモジュールは、側鎖原子の実際の誤差 (生成された座標と実際の座標間の距離) を 33 の区間に分割します。このモデルは、カテゴリカルクロスエントロピー損失を用いて学習され、生成過程の情報に基づいて各原子エラーの間隔を予測し、間隔確率の期待値を通じて最終的なエラー推定値pSCEを得ることができます。このモジュールの入力には、全原子エンコーダの特性、生成された配列と側鎖の座標、拡散過程におけるノイズレベルが含まれます。出力pSCEは側鎖パッキングの精度を効果的に反映し、高品質な設計結果の選別とモデルの解釈可能性の向上に役立ち、側鎖構造生成の品質評価リンクを改善します。
実験結果: メインチェーンのみに基づくモデルよりもパフォーマンスが大幅に向上しました
モデルの性能を検証し、正確に評価するために、研究チームはまず、配列回復と自己一貫性の評価実験を行い、FAMPNNを他の手法と比較しました。具体的な比較対象は下図に示されています。
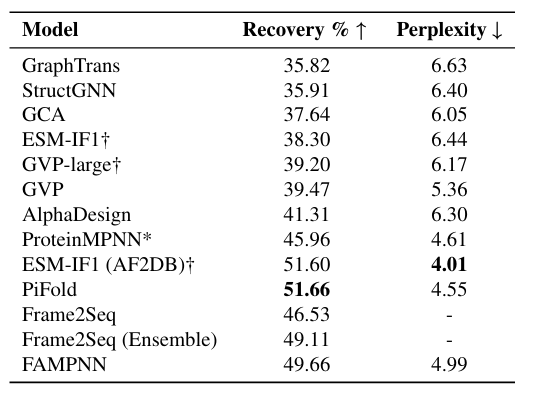
実験によればFAMPNN は、シングルステップのシーケンス回復精度に関して現在の最先端の方法を上回り、49.66% に達します。比較すると、ProteinMPNN はわずか 45.96%、GVP はわずか 39.47% です。
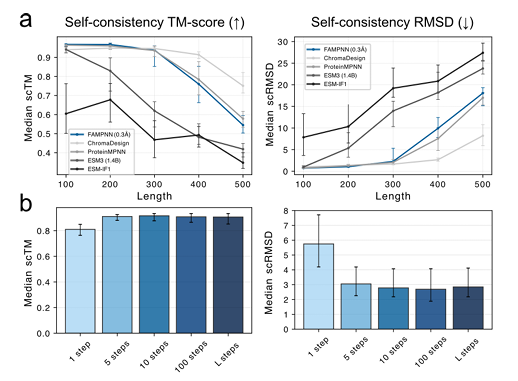
RF拡散に基づいて生成された新しい主鎖の自己一貫性評価では、FAMPNN (0.3Å) は、scTM (構造類似性) および scRMSD (二乗平均偏差) メトリックの点で ProteinMPNN に匹敵します。10段階の反復サンプリングにより、高い自己一貫性を実現でき、これは完全な自己回帰法よりも効率的です。次の図をご覧ください。
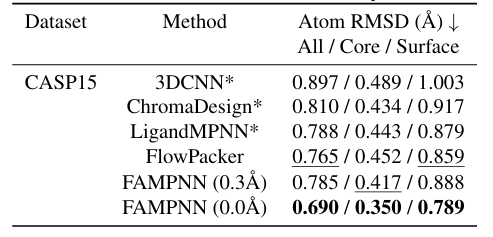
サイドチェーンパッキングに関しては、研究者らは提案モデルを CASP13、14、15 データセット上の他の方法と比較しました。実験では、CASP15 テスト セットの結晶構造評価において、FAMPNN (0.0Å) の原子 RMSD (全体/コア/表面) は 0.690/0.350/0.789Å となり、他の方法よりも優れていることが示されています。また、各原子の誤差および各残基の誤差と強い相関関係があり、スピアマン相関係数はそれぞれ0.843と0.780です。下の図をご覧ください。
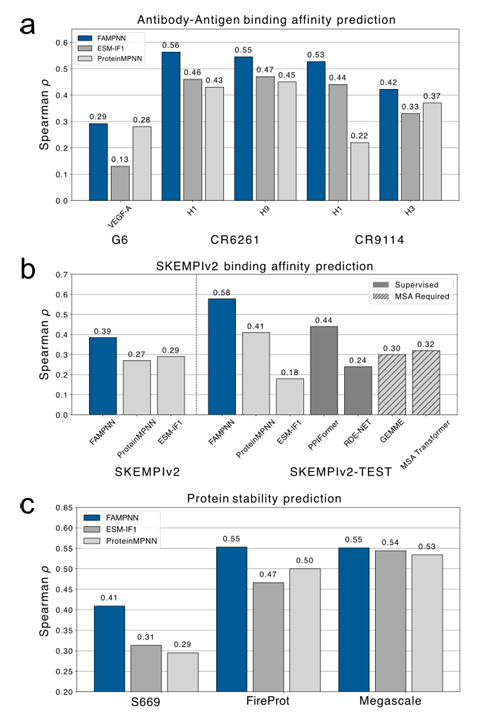
全原子条件下でのタンパク質の適応度評価では、SKEMPlv2データセットでは、FAMPNNは教師なしモデルを大幅に上回り、テストサブセットでは教師ありモデルさえも上回り、ゼロサンプル予測において高い一般化能力を示しました。S669、Megascale、FireProtDBの3つの安定性データセットでは、FAMPNNはProteinMPNNとESM-IFをわずかに上回りました。抗体-抗原結合親和性の予測において、FAMPNNは常に最先端の教師なし手法であるProteinMPNNとESM-IF1を上回り、タンパク質の安定性とタンパク質間相互作用の強化におけるFAMPNNの有用性を証明しました。次の図をご覧ください。
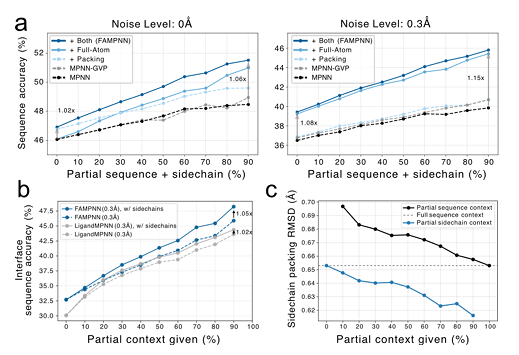
全原子モデリングが配列設計のパフォーマンスを向上させるかどうかを評価する実験では、研究では、側鎖パッキングターゲットと全原子条件設定を追加すると、配列精度が向上することがわかりました。さらに、FAMPNNとベースラインモデルの両方の性能は、より多くの構造情報が注入されるにつれて向上します。タンパク質-タンパク質界面では、側鎖相互作用のモデリングがより重要になり、配列と併せて部分的な側鎖コンテキストを提供することで、部分的な配列コンテキストのみを提供する場合と比較して、精度を大幅に向上させることができます。
さらに、LigandMPNNと比較して、FAMPNNは側鎖コンテキストをより効果的に活用し、部分配列または側鎖コンフォメーションコンテキストの数に基づいて側鎖パッキングを実行できます。コンテキストの数が多いほど、パッキング精度が向上します。下の図cに示されています。
要約すると、上記の実験は、メインチェーンのみに基づくモデルと比較して、FAMPNN がタンパク質の適応度予測において大きな利点があることを示しています。
人工知能の推進により、学術界はサイドチェーンモデリングの分野で繁栄している
冒頭で述べたように、側鎖の立体配座はタンパク質の機能にとって極めて重要です。しかし、タンパク質の主鎖が決定された後も、側鎖の立体配座は依然として多くの可能性を秘めており、側鎖立体配座のモデリングと研究は克服すべき困難な課題となっています。本研究に加え、世界中の多くの学術研究機関が、最先端のディープラーニング技術と生物学的知見を活用し、側鎖モデリングの研究を推進しています。
中国の復旦大学のチームは、OPUS-Rota5と呼ばれる2段階側鎖モデリング法を提案しました。この方法では、改良された 3D-Unet を使用してローカル環境の特徴をキャプチャします。各残基のリガンド情報を取り込み、RotaFormerモジュールを用いて様々な特徴量を集約します。CAMEOやCASP15などのテストセットを用いた評価では、OPUS-Rota5が他の主要な側鎖モデリング手法を大幅に上回る性能を示すことが示されました。関連研究は、「OPUS-Rota5:3D-UnetとRotaFormerを用いた高精度タンパク質側鎖モデリング手法」というタイトルでScienceDirectに掲載されました。
用紙のアドレス:
https://www.sciencedirect.com/science/article/pii/S0969212624001266
北京大学のチームは、GeoPackerと呼ばれる別の方法を提案しました。この手法では、幾何学的ディープラーニングと ResNet を組み合わせて、タンパク質側鎖をモデル化します。 GeoPackerは、回転および並進不変な方法で原子間相互作用を明示的に表現し、相対位置情報を抽出します。側鎖構造予測の精度において、GeoPackerはエネルギー関数に基づく最先端の手法を凌駕し、ディープラーニングベースの手法であるDLPackerおよびOPUS-Rota4と比較して、それぞれ約10倍、約700倍の速度で、同等の予測精度を実現します。関連研究は「GeoPacker:タンパク質側鎖モデリングのための新たなディープラーニングフレームワーク」として発表されました。
用紙のアドレス:
https://onlinelibrary.wiley.com/doi/epdf/10.1002/pro.4484
同時に、トロント大学のチームはFlowPackerと呼ばれるモデルを提案しました。その目的は、タンパク質の既知のアミノ酸配列と主鎖構造に基づいて、側鎖の特定の形状を正確に予測することです。FlowPackerは、従来の高度な手法と比較して、ほとんどの指標において優れたパフォーマンスを発揮し、実行速度も向上しています。例えば、角度予測誤差、予測角度と真値の近似度、原子位置の偏差などにおいて優位性があります。関連研究は「FlowPacker:ねじれフローマッチングを用いたタンパク質側鎖パッキング」というタイトルで発表されています。
用紙のアドレス:
一般的に、側鎖構造の解読は生命科学分野の発展にとって極めて重要です。人工知能技術の継続的な発展は、構造生物学と計算生物学の急速な発展を間違いなく促進し、国内外の研究機関が学術的成果を飛躍的に向上させることに貢献してきました。これらの成果が実験室から応用へと応用されれば、生命科学分野に新たな旋風を巻き起こし、生物学と医学を新たな時代へと導くことは間違いありません。








