Command Palette
Search for a command to run...
MITとハーバード大学は、タンパク質言語モデルと画像修復モデルを統合し、単一細胞タンパク質の局在化を実現するPUPSを共同で提案した。
タンパク質の細胞内局在とは、細胞構造におけるタンパク質の特定の位置を指します。これはタンパク質が生物学的機能を果たすために不可欠です。簡単な例を挙げると、細胞を巨大な企業体とみなし、細胞核、ミトコンドリア、細胞膜などがそれぞれ社長室、発電部門、門番などの各部署に相当し、対応するタンパク質が正しい「部署」に入って初めて正常に機能することができ、そうでなければ、がんやアルツハイマー病などの特定の疾患を引き起こします。したがって、タンパク質の正確な細胞内局在の特定は、生命科学の中核課題の 1 つであると言えます。
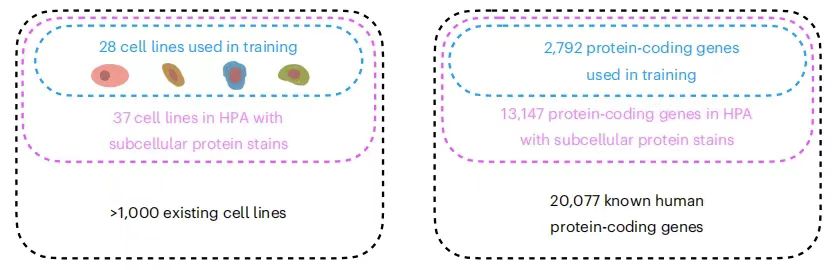
さまざまな細胞株における何千ものタンパク質の空間的局在が分析されているものの、これまでに測定されたタンパク質と細胞株の組み合わせの数は氷山の一角にすぎません。たとえば、現在利用可能な最大の細胞内局在データセットは次のとおりです。ヒトタンパク質アトラス (HPA) は、13,147 個の遺伝子 (既知のヒトタンパク質コード遺伝子の 65% を占める) によってコード化されたタンパク質の細胞内局在を提供します。ただし、データセット全体には 37 個の細胞株が含まれており、各タンパク質は最大 3 つの細胞株で測定されていました。同時に、主流の実験方法では、同じ細胞内のすべてのタンパク質の数を同時に検出することが困難であり、複雑なタンパク質ネットワークの包括的な分析を著しく妨げ、実験の複雑さとエラーのリスクが増大します。
さらに、タンパク質の局在は静的ではなく、細胞株間だけでなく、同じ細胞株内の個々の細胞間でも変動が生じます。既存のデータ マップに記録されたタンパク質と細胞株のペアは、特定の条件下での結果のみを反映します。したがって、既存の結果であっても直接適用することは難しく、環境の変化に基づいたタンパク質の局在のさらなる探究が必要です。
タンパク質細胞内局在技術手法の限界と生物システムの複雑性との間の矛盾を解決するために、機械学習が有望であると期待されています。タンパク質配列ベースのモデルや細胞画像ベースのモデルなど、現在構築され、うまく適用されているモデルは、いくつかの面では優れたパフォーマンスを発揮していますが、その欠点も非常に顕著です。前者は細胞タイプの特定の局在の違いを無視し、後者は未知のタンパク質の研究を促進するための一般化能力を欠いています。
これを踏まえて、マサチューセッツ工科大学とハーバード大学の研究チームは、タンパク質配列と細胞画像を組み合わせることで未知のタンパク質の細胞内局在を予測するフレームワーク「Predictions of Unseen Proteins' Sub-cellular localization (PUPS)」を提案した。 PUPS は、タンパク質言語モデルと画像修復モデルを革新的に組み合わせてタンパク質の局在を予測し、未知のタンパク質予測の一般化機能と、細胞の変動性を捉える細胞タイプ固有の予測を統合します。実験により、このフレームワークはトレーニングデータセット外の新しい実験でタンパク質の局在を正確に予測でき、優れた一般化能力と高い精度を備え、優れた応用可能性を秘めていることが示されました。
「単一細胞におけるタンパク質の細胞内局在の予測」と題された研究成果は、Nature Methods誌に掲載されました。
研究のハイライト:
* 提案された研究は、タンパク質言語モデルと画像レンダリングモデルを革新的に組み合わせ、タンパク質配列と細胞画像を使用してタンパク質の局在を予測し、従来の計算モデルの欠点を補うものである。
* PUPSは未知のタンパク質や細胞株に一般化できるため、細胞株間および細胞株内の個々の細胞間のタンパク質局在の変動性を評価し、変動する局在を持つタンパク質に関連する生物学的プロセスを特定することができます。
* トレーニングデータセット以外の新しい実験では、PUPSは非常に正確な予測能力を示し、優れた応用可能性と医学的価値を備えていることが示されました。

用紙のアドレス:
データセット: 可能な限り包括的なデータを使用して信頼できるモデルを構築する
PUPS のトレーニング データ セットは、Human Protein Atlas (HPA) から取得されます。研究チームは、できるだけ多くのタンパク質データを収集し、実験分析の包括性を確保するために、HPA データの第 16 バージョンを第 22 バージョンに集約しました。次の図に示すように:
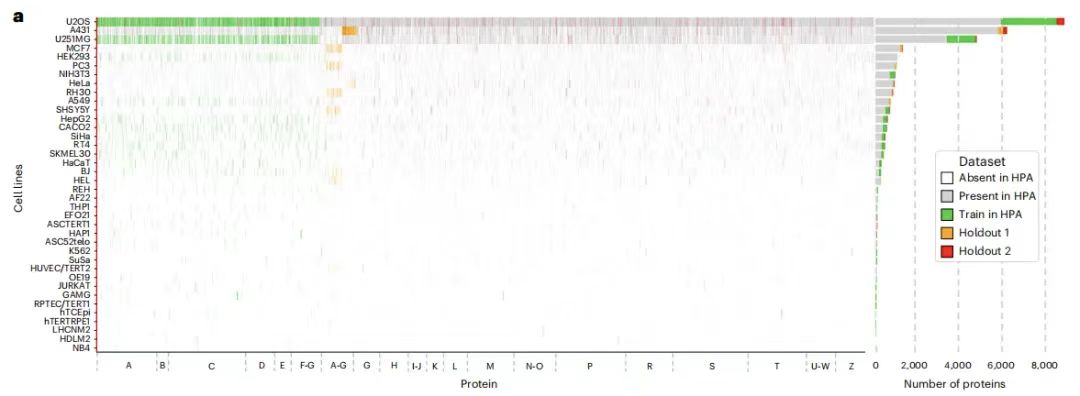
具体的には、トレーニング データセットには、HPA の 37 個の細胞株の 2,801 個の遺伝子に対応する、合計 8,086 個のタンパク質変異体を持つ 340,553 個の細胞集団が含まれており、名前は AG の文字で始まります。さらに、トレーニング データセットには、IHO1、IMPAD1、INKA1、ISPD、ITPRID1、KIAA1211L、KIAA1324、LRATD1、SCYL3、TSPAN6 を含む 10 個の追加遺伝子も含まれています。
ホールドアウト データセットは 2 つの部分に分かれています。1つの部分は予約データセット1です。このデータセットには 36,552 個のセルが含まれており、3,312 個の遺伝子 (トレーニング セット内の 2,801 個を含む) に対応する 9,472 個のタンパク質変異体が含まれています。これらの遺伝子の名前も AG で始まりますが、異なる細胞株からのものであり、トレーニング セットとは重複しません。一方、保留データセット 1 はさらに 2 つの部分に分割され、それぞれ 11,050 個と 25,502 個のセルを含む評価セットとテスト セットとして使用されました。保持されたデータセット 2 には、515 個の遺伝子に対応する 24,007 個のセルが含まれています。その名前はアルファベットのすべての文字で始まり、AZ をカバーします。合計 556 個のタンパク質変異体があり、これらはトレーニング セットや予約済みデータセット 1 には表示されない新しい遺伝子ファミリーに由来し、モデルの一般化能力をテストするために使用できます。
BJ 細胞株画像はトレーニング セットとホールドアウト データセット 1 の両方で保持されていることに注意してください。
実験の前に、研究チームは HPA で画像を前処理しました。これは次の 5 つの手順だけで済みます。
* ステップ1、計算量を削減し、高周波ノイズを除去するために、各画像は 4 回ダウンサンプリングされ、最終解像度は 0.32 μm/ピクセルに削減されました。
* ステップ2、ガウスぼかし(σ=5)とOtsu閾値処理を組み合わせて、細胞核のおおよその領域を複雑な背景から分離しました。
* ステップ3、remove_small_holes 関数を使用して、面積が 300 ピクセル未満の穴を削除し、画像を 2 値化して、100 ピクセル未満のノイズ領域を削除します。
* ステップ4、各細胞核の重心が計算され、その重心が単一細胞の ROI として 128 × 128 ピクセルの領域が切り取られました。
* ステップ5、強度の正規化とノイズフィルタリングにより、標準化されたデータ分散が実現され、チャネル間干渉が削減されます。
モデルアーキテクチャ: タンパク質配列と画像表現を組み合わせてタンパク質の細胞内局在を予測する
PUPS モデルは主に 2 つの部分で構成されます。1 つは、タンパク質のアミノ酸配列から配列表現を学習するために使用されます。もう 1 つは、ターゲット セルの象徴的な染色から画像表現を学習するために使用されます。次に、タンパク質配列表現と画像表現を組み合わせて、標的細胞におけるタンパク質の細胞内局在を予測します。前者はモデルを未知のタンパク質予測に一般化することを可能にし、後者はモデルが単一細胞レベルで変動を捉え、細胞タイプ固有の局在予測を達成することを可能にします。次の図に示すように:
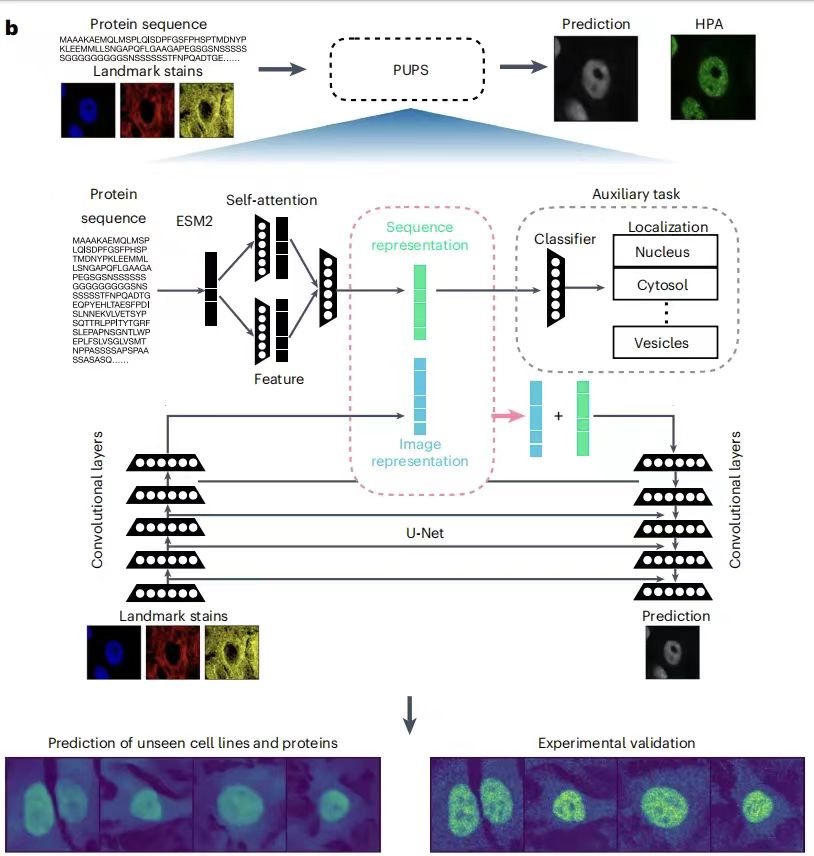
簡単に言えば、PUPS は、事前トレーニング済みの ESM-2 (Evolutionary Scale Modeling) タンパク質言語モデルを使用してタンパク質配列の特徴を抽出し、畳み込みニューラル ネットワークを使用して細胞の象徴的な染色画像の特徴を学習します。最後に、2 つの情報を組み合わせて、標的細胞におけるタンパク質の局在を予測します。モデルのすべての部分が同時にトレーニングされる点に留意する必要があります。これにより、事前タスクの分類損失と、HPA で予測されたタンパク質画像と実験的に測定されたタンパク質画像との差が削減されます。すべてのパラメータは、学習率 1e-4 の Adam オプティマイザーを使用して最適化されます。
タンパク質言語モデル
PUPS は、言語モデル、自己注意層、および補助的な事前トレーニング タスクを使用してシーケンス表現を学習し、学習したシーケンス表現に基づいてタンパク質の局在を分類します。
具体的には、研究チームは、事前学習済みの ESM-2 モデルに N 末端の 2,000 アミノ酸配列を入力することで特定のタンパク質変異体の初期表現を取得し、アミノ酸残基ごとに 1,280 次元のベクトルを生成しました。2,000 残基未満の変異体についてはパディングをゼロにしました。この配列長のカットオフは、配列長が最大数万残基に達する少数のタンパク質に対する偏った予測を回避することを目的としています。次の図に示すように:
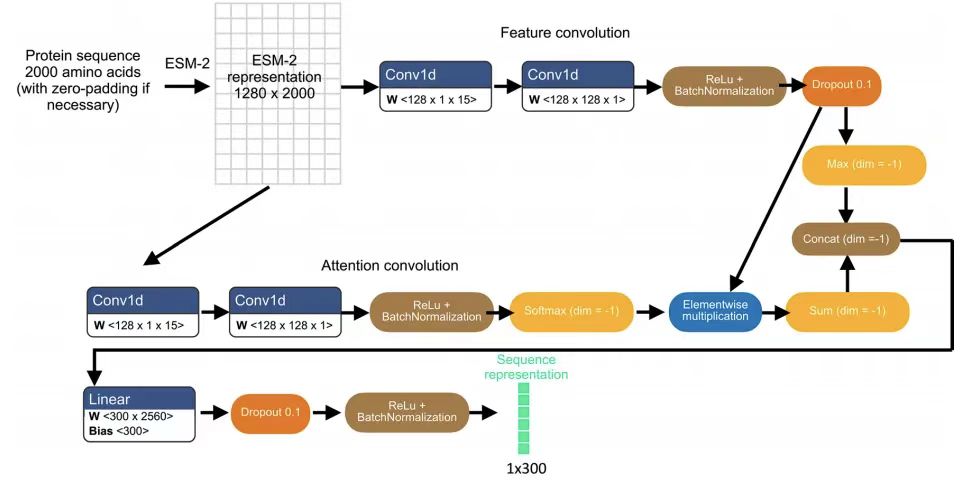
ESM-2の特性をタンパク質の局在予測に適応させるには、その後、チームは分離可能な畳み込みの軽い注意層を採用しました。ESM-2 表現に適用すると、最終的に 300 次元のシーケンス表現が得られます。このタンパク質配列表現は、局在ラベルを予測する補助的な事前タスクと、画像表現と組み合わせたタンパク質画像予測の両方に使用されます。事前タスクでは、タンパク質配列表現を完全接続ニューラル ネットワーク層に入力して、29 個の細胞内コンパートメントの局在ラベル間の確率分布を表す 29 次元ベクトルを入力し、次に、シグモイド活性化によるバイナリ クロスエントロピー損失を使用して、事前タスク出力を HPA 注釈付きタンパク質コンパートメントと比較します。
画像レンダリングモデル
各細胞の画像入力には、細胞核、微小管、小胞体染色の 3 つの象徴的な染色画像チャネルが含まれています。その寸法は 3 x 128 x 128 で、核の重心を中心としています。
画像のエンコードは、5 つの分離可能な畳み込み層を通じて実現されます。最終寸法 16 x 16 x 512 。各畳み込み層の後には、leakyRelu アクティベーション、バッチ正規化、および 2D 最大プーリング層が続きます。タンパク質配列表現は、細胞画像表現のすべての空間次元に連結され、入力チャネルごとに異なる重みを学習する U-Net 画像デコーダーに送られます。さらに、モデルの空間次元重み付けメカニズムにより、画像表現の各空間次元を異なる重みでシーケンス表現と組み合わせることができます。
デコーダーは 5 つの分離可能な畳み込み層で構成されています。対応するセルのタンパク質画像予測である 1 x 128 x 128 の画像出力を生成します。次に、ランドマーク染色の画像表現を生成するエンコード層と、同じ深さでタンパク質画像予測を生成するデコード層の間に、画像セグメンテーション U-Net と同様のスキップ接続が追加されます。この研究では、平均二乗誤差損失関数を使用してモデルをトレーニングし、予測されたタンパク質画像と実験的に測定されたタンパク質画像との差を最小限に抑えました。
実験結果:単一細胞レベルでのタンパク質の正確な細胞内局在の実現
モデルの実現可能性と有効性を検証するために、研究チームはいくつかの検証実験を提案しました。 PUPS は複数のタスクで優れたパフォーマンスを示し、マルチモデル融合における利点を強調しました。
細胞株間のタンパク質局在の変動を予測する
細胞株間のタンパク質局在の変動を定量化するPUPSの性能を評価するために、研究チームはタンパク質核比率を計算して局在の変動性を定量化し、予測値が実際のデータと高い相関関係にあることを発見した。ホールドアウト 1 のピアソン相関係数は 0.794 で、ホールドアウト 2 のピアソン相関係数は 0.878 です。次の図に示すように:
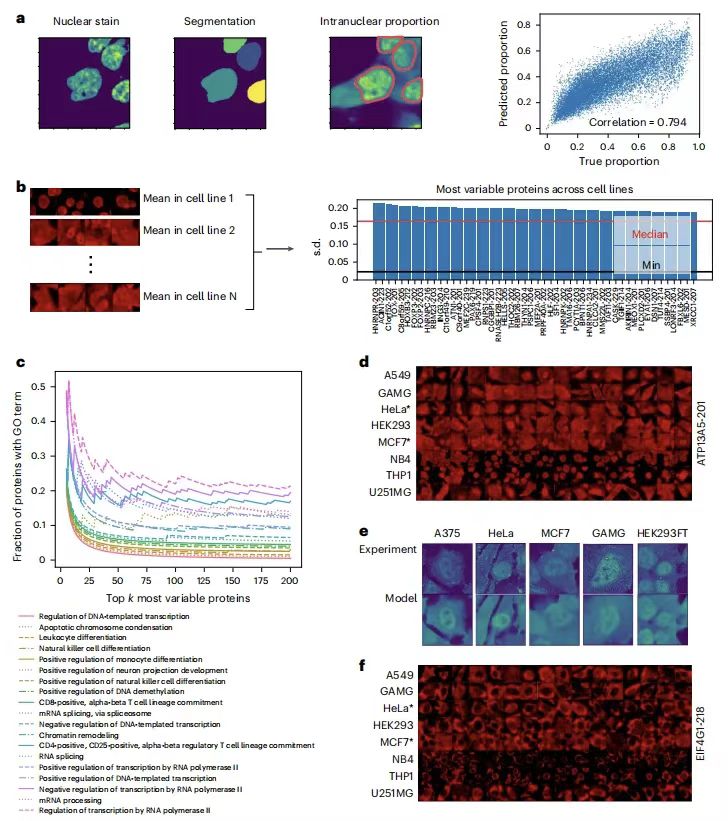
その後のさらなる分析により、細胞株間で最も大きな局在変化を示すタンパク質は、転写、細胞分化、クロマチン調節などの生物学的プロセスに関連していることが判明しました。 ATP13A5 の実験的検証により、モデルの予測の正確性が確認されました。また、このモデルは、シグネチャー染色を通じて細胞形態の違いを捉え、細胞株ラベルなしでタンパク質の局在の細胞株特異性を推測することができる。細胞特異的なタンパク質機能の調節を研究するための新しい方法を提供します。
単一細胞間のタンパク質局在の違いを予測する
同じ細胞株内の単一細胞間のタンパク質局在の変動を予測するPUPSの能力を評価するために、研究チームは各細胞株内のすべての単一細胞におけるタンパク質の核比率の分散を計算しました。結果は、各タンパク質と細胞株のペアの単一細胞変動予測ランキングが実際のデータと非常に一致していることを示しました。例えば、Holdout 2 では、最初の 500 個の高変異ペアの重複率が 60% を超えており、予測された核内比率の分布は実際の結果と一致しており、予測誤差の影響が排除されています。
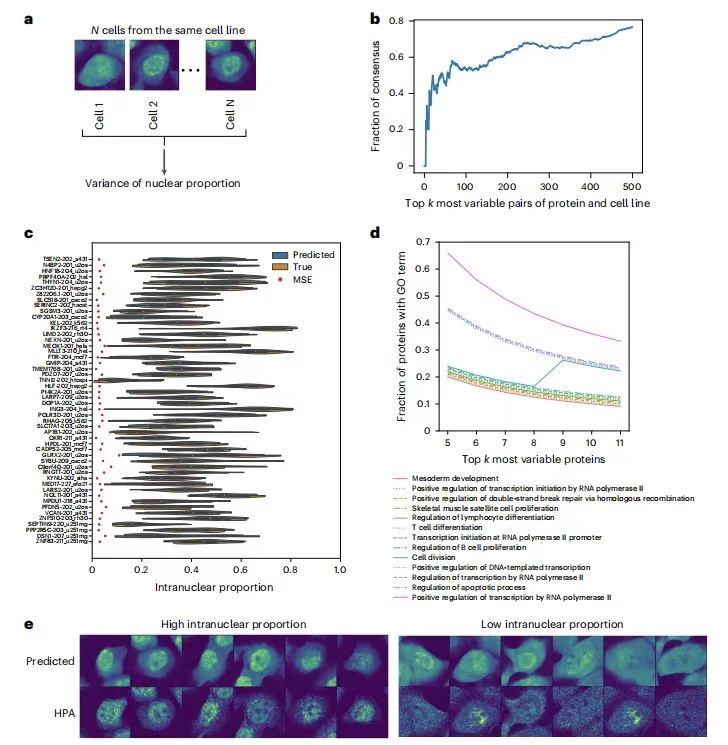
さらに、遺伝子オントロジー (GO) 分析により、非常に変動の大きいタンパク質が細胞分裂、転写、二本鎖切断修復、アポトーシスなどのプロセスに関連していることが示されました。また、このモデルは、細胞マーキング染色画像を通じて形態学的特徴を捉え、単一細胞の変動がランダムであるだけでなく、細胞の形態学的特徴にも関連していることを示しています。単一細胞の異質性のメカニズムを説明するための新しい視点を提供します。
トレーニングデータ以外の新しい実験におけるPUPSの検証
新たな実験環境におけるタンパク質の局在を予測する PUPS のユビキチン化能力を検証するために、研究チームは 5 つの細胞株で検証用に 9 つのタンパク質を選択しました。次の図に示すように:
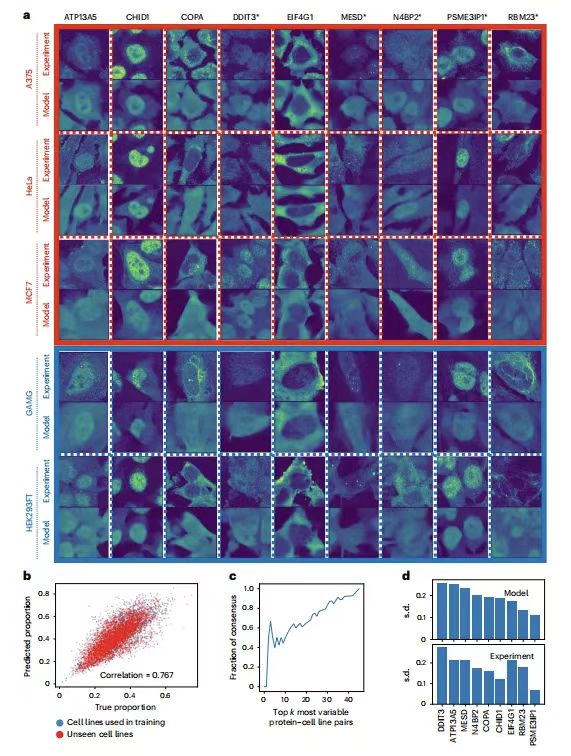
ATP13A5、CHID1、COPA、MESD、RBM23 は細胞株間で最も大きな変異を持つタンパク質であり、それらはすべて異なる GO 用語を持ちます。 DDIT3 と N4BP2 は、細胞株内の個々の細胞で最も大きな変異を持つタンパク質です。 EIF4G1 と PSME3IP1 は細胞株間での変異が最も少ないタンパク質であり、前者は主に核の外側に位置し、後者は主に核の内側に位置すると予想されます。 5 つの細胞株のうち、A375 を除く他の HeLa、MCF7、GAMG、および HEK293FT は HPA に含まれます。
結果は次のようになります。PUPS によって予測されたタンパク質画像は、実験的に測定されたものと視覚的に類似しています。予測タンパク質画像を使用して計算された各単一細胞の核タンパク質比率は、実験的に測定された画像から計算された比率と密接に相関しており、ピアソン相関係数は 0.767 です。これは、PUPS は、これまで実験的に測定されたことも、トレーニング アトラスで使用されたこともないタンパク質の局在を定量的に予測するために使用できます。
PUPSは意味のあるタンパク質と細胞の表現を学習します
実験により、PUPS が未知のタンパク質や細胞株におけるタンパク質の局在を予測する能力は、タンパク質配列と細胞ランドマーク画像の意味のある表現を学習することから生まれることが実証されています。
研究チームは、12,614 個の遺伝子に対応する 40,622 個のタンパク質形態のタンパク質配列表現をマッピングしたところ、類似した局在を示すタンパク質は類似した配列表現を示す傾向がありました。モデルが意味のあるタンパク質配列パターンを識別し、局在を予測できることをさらに実証するために、研究チームは、位置シャプレー法を使用して、特定のタンパク質内の各アミノ酸残基の重要性を計算し、各細胞区画のラベルを予測しました。たとえば、N4BP2 の核局在の予測される変動性をうまく説明することができ、これは CUE ドメインがユビキチン結合を通じて細胞内局在を変化させる可能性があるという報告と一致しています。
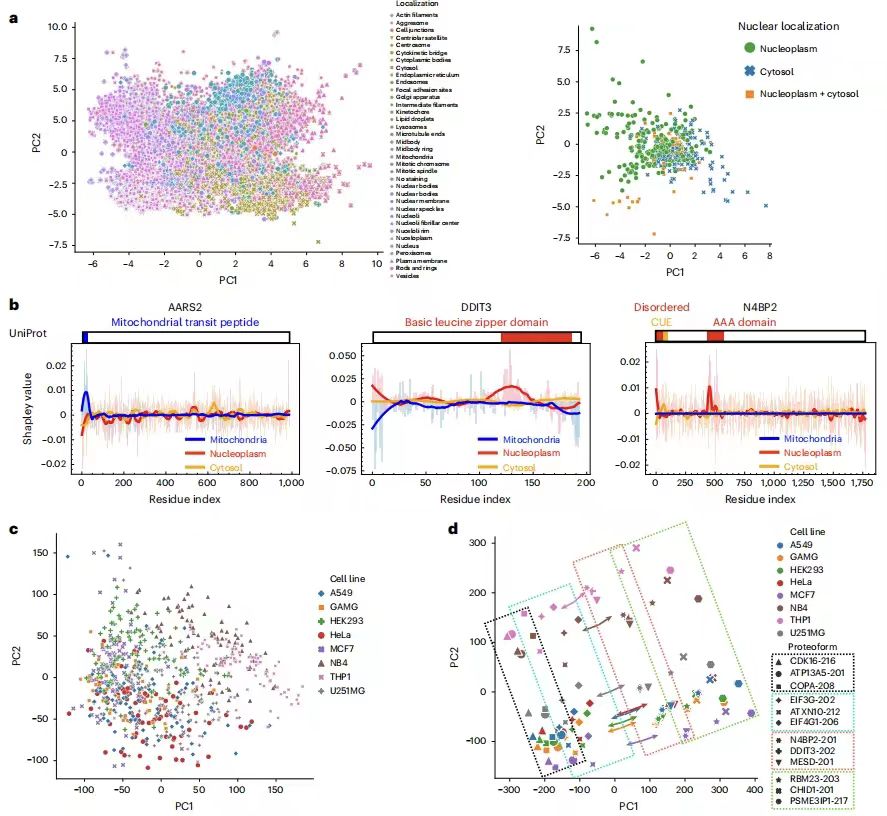
意味のあるタンパク質配列モチーフを特定することに加えて、研究チームはさらに、PUPS が細胞シグネチャー染色から単一細胞の意味のある表現を学習することを実証しました。ランドマーク染色から学習した単一細胞画像表現を視覚化し、細胞株ラベルがモデルに入力されていない場合でも、同じ細胞株の単一細胞が同様の画像表現を持つことを発見します。タンパク質と細胞ランドマーク画像の結合表現により、細胞株とタンパク質の分離が維持されますが、各細胞株内の異なるタンパク質の順序は細胞株間で同様になります。共同表現空間内の各細胞株の重心が与えられれば、重心から特定のタンパク質へのベクトルはすべての細胞株にわたってほぼ平行になります。つまり、配列表現が与えられれば、特定のタンパク質の画像を予測するには、細胞株に関係なく、表現空間内で同じ方向に移動する必要があります。これは、タンパク質と細胞の画像の意味のある表現を学習することにより、PUPS が未知のタンパク質と細胞株に一般化できる能力を説明しています。
また、PUPS は、疾患の原因となる変異がタンパク質の局在に及ぼす影響を予測することもできます。例えば、核コード化ミトコンドリアタンパク質 SDHD および ETHE1 の変異研究では、SDHD 変異によりそれらの核局在率の増加がもたらされることが示されており、これは疾患における核ゲノム不安定性のメカニズムと一致しています。 ETHE1 変異は細胞質局在比の増加を示し、これは既知の核-細胞質シャトル異常と関連しています。これらの結果は、PUPS が配列変異の局在に対する影響を分析することによって疾患メカニズムの研究に新たな手がかりを提供できることを示唆しています。
タンパク質の細胞内局在予測のための新しいソリューション
前述のように、タンパク質の細胞内局在予測は、バイオインフォマティクスと生物学研究の両方において非常に重要です。 PUPS は、マルチモーダル情報を統合する方法を提供し、この分野の研究に大きなインパクトを与えます。同時に、数十年にわたる発展を経て、この分野の研究は多種多様な成果を生み出してきました。
アイルランドのダブリン大学カレッジのチームが「Computational and Structural Biotechology Journal」に研究論文を発表した。シーケンスベース、アノテーションベース、ハイブリッド、メタ予測ベースの方法など、タンパク質の細胞内局在予測のためのさまざまな計算方法を紹介しています。また、この記事では、真核生物、原核生物、ウイルスと複数のカテゴリ別に細胞内局在予測ツールを分類して紹介しています。真核生物の予測ツールにはmLASSO-Hum、DeepPSLなどがあり、原核生物の予測ツールにはPRED-LIPOなどがあります。本研究では、7つの主要領域と28のサブカテゴリをカバーする機械学習とディープラーニングの分類マップを設計することにより、単一カテゴリと複数カテゴリの予測ツールの分類を提供し、ユーザーが方法と予測ツールを見つけやすくしました。この論文は「タンパク質細胞内局在予測ツール」として発表されました。
* 紙のアドレス:
https://www.sciencedirect.com/science/article/pii/S2001037024001156
4月12日、復旦大学生物医学研究所の楊立氏の研究グループと上海人工知能研究所の董南青氏の研究グループが協力し、「タンパク質細胞内局在のための深層生成モデル」と題するオンライン研究論文をBriefings in Bioinformatics誌に発表した。この研究では、ESM2 タンパク質言語モデルと U-Net フレームワークに基づくマルチモーダル処理機能を備えた生成ディープラーニング モデル deepGPS も開発されました。
報道によれば、deepGPSはタンパク質配列や細胞核画像を入力として受け取り、タンパク質の局在を示すテキストラベルや分布画像を生成できるという。これは、タンパク質の細胞内局在の予測をサポートする新しい「テキストから画像への」マルチモーダル モデルです。
* 紙のアドレス:
https://doi.org/10.1093/bib/bbaf152
人工知能と生物学研究の統合が加速するにつれて、関連する革新的な実験が絶えず登場し、徐々に従来の方法の欠点を打ち破り、「両方の長所を兼ね備えた」、あるいは「完璧な」パフォーマンスを実現し、バイオインフォマティクスの急速な発展を促進しています。








