Command Palette
Search for a command to run...
ウェストレイク大学のチームは、構造機能予測/クロスモーダル情報検索/アミノ酸配列設計などをカバーする SaProt およびその他のタンパク質言語モデルをオープンソース化しました。

2025年3月22日〜23日、上海交通大学の「AIタンパク質設計サミット」が正式に開催されました。サミットには、清華大学、北京大学、復旦大学、浙江大学、厦門大学などの有名大学から300人以上の専門家や学者、さらに業界大手企業や技術研究開発担当者から200人以上の代表者が集まり、タンパク質設計分野におけるAIの最新の研究成果、技術革新、産業応用の見通しについて深く議論しました。

サミット中、西湖大学の袁法傑博士は、「タンパク質言語モデルの研究と応用」をテーマに、タンパク質言語モデルの最新の研究進捗状況を共有し、チームの重要な成果を詳しく紹介しました。タンパク質言語モデルSaProt、ProTrek、Pinal、Evollaなどを含め、HyperAIは本来の意図を損なわずに詳細な共有を整理し、まとめました。以下はスピーチのハイライトの書き起こしです。
注目に値するタンパク質言語モデル
タンパク質は、20 個のアミノ酸が連続して結合して構成される生物学的高分子です。体内で触媒や代謝などの重要な機能を果たし、生命活動の主な実行者です。生物学者は通常、タンパク質の構造を 4 つのレベルに分類します。一次構造はタンパク質のアミノ酸配列を記述し、二次構造はタンパク質の局所的な立体配座に焦点を当て、三次構造はタンパク質の全体的な 3 次元構成を表し、四次構造は複数のタンパク質分子間の相互作用に関係します。AIタンパク質の分野では、主にこれらの構造に基づいた研究が行われています。
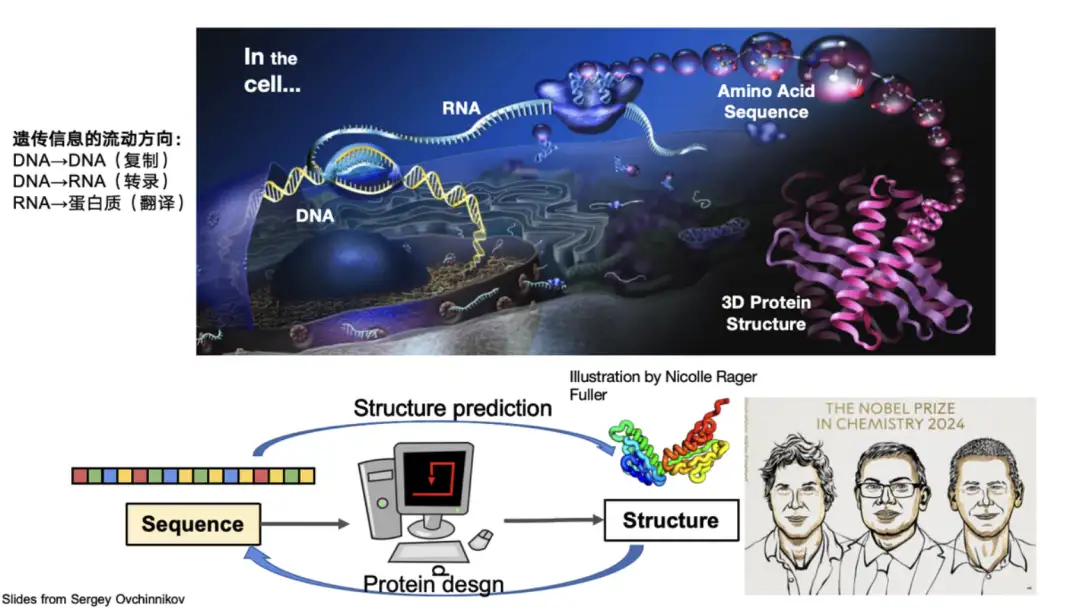
たとえば、タンパク質の配列からその 3 次元構造を予測することが、AlphaFold 2 が解決する中心的な問題です。科学界を50年間悩ませてきたタンパク質の折り畳み問題を克服し、ノーベル賞を受賞した。一方、構造と機能に基づいて新しいタンパク質配列を設計するタンパク質設計の分野で重要な貢献をしたデビッド・ベイカー教授もノーベル賞を受賞しました。
伝統的に、タンパク質構造は通常、PDB 座標の形式で表されます。近年、研究者は、Foldseek、ProTokens、FoldToken、ProtSSN、ESM-3 など、連続的な空間構造情報を離散的なトークンに変換する方法を研究してきました。
*Foldseek は、タンパク質の 3 次元構造を 1 次元の離散トークンにエンコードできます。
私たちのチームのタンパク質言語モデルは、これらの個別の結果に基づいています。
AI + タンパク質研究のほとんどは自然言語処理研究に遡ることができるので、まずは自然言語処理 (NLP) の分野における 2 つの古典的な言語モデルを見てみましょう。1つはGPTシリーズに代表される一方向言語モデルであり、そのメカニズムは左から右への情報の流れに基づいており、左側(上側)のデータに基づいて次のトークンを予測します。1つはBERTに代表される双方向言語モデルであり、これは、Masked Language Model を通じて事前トレーニングされており、調理された単語の左側と右側の情報 (コンテキスト) を確認し、調理された単語を予測することができます。
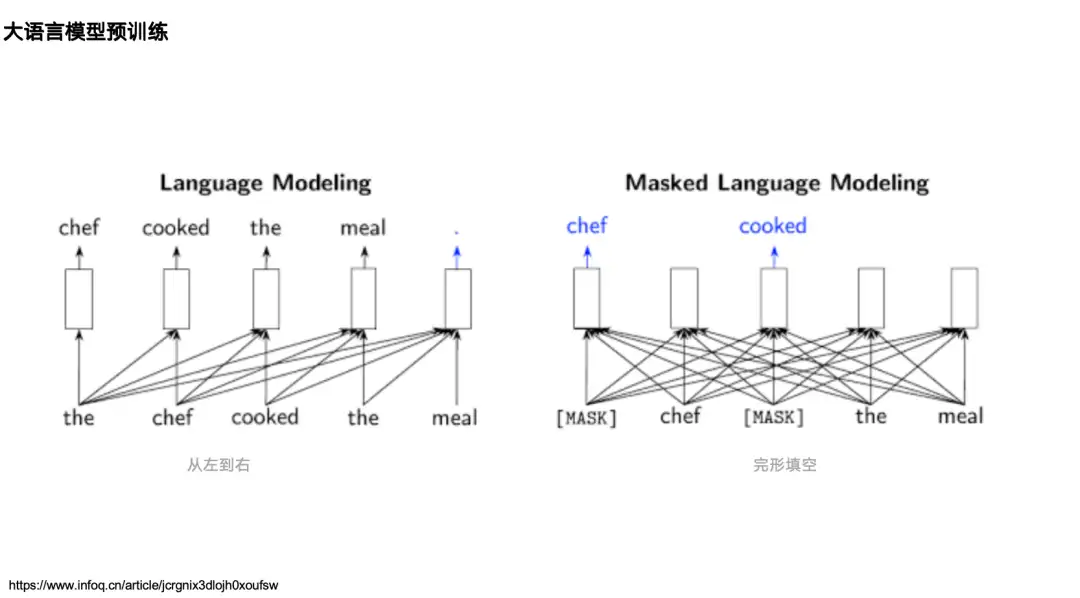
タンパク質分野では、両方のタイプのモデルに対応するタンパク質言語モデルがあります。例えば、GPT に対応するものとしては、ProtGPT2、ProGen などがあります。BERT に対応するものとしては、ESM シリーズ モデル ESM-1b、ESM-2、ESM-3 があります。それらは主にいくつかのアミノ酸をマスクし、その「真の正体」を予測します。自然言語タスクでは、いくつかの単語をマスクして予測します。下の図の左側に示すように、タンパク質コミュニティで比較的大きな影響力を持つ他の言語モデルとしては、MSA Transformer、GearNet、ProTrans などがあります。
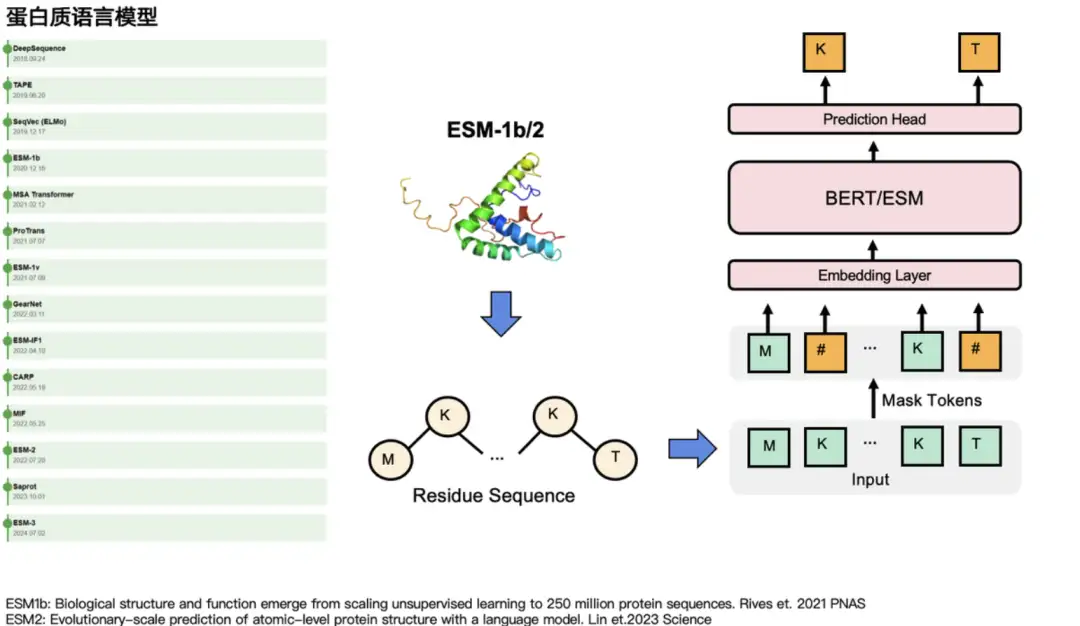
ICLR 2024に選ばれたタンパク質言語モデルSaProtは構造知識を統合します
最初にご紹介したい成果は、構造を考慮した語彙を備えたタンパク質言語モデルである SaProt です。「SaProt: 構造を考慮した語彙によるタンパク質言語モデリング」と題されたこの論文は、ICLR 2024 に選ばれました。
本論文では、構造を考慮した語彙の概念を提案し、アミノ酸残基トークンと構造トークンを組み合わせ、約 4,000 万のタンパク質配列と構造のデータセットで大規模な汎用タンパク質言語モデル SaProt をトレーニングしました。このモデルは、10 個の重要な下流タスクにおいて、既存の成熟したベースライン モデルを総合的に上回りました。
SaProt オープンソース アドレス:
https://github.com/westlake-repl/SaProt
SaProt 紙のアドレス:
https://openreview.net/forum?id=6MRm3G4NiU
なぜこのモデルを作るのでしょうか?
実際、ほとんどのタンパク質言語モデルの入力情報は主にアミノ酸配列に基づいています。 AlphaFold での画期的な進歩の後、DeepMind チームは欧州バイオインフォマティクス研究所 (EMBL-EBI) と協力し、2 億個のタンパク質構造を保存する AlphaFold タンパク質構造データベースをリリースしました。そこで私たちは考え始めました。タンパク質の構造情報を言語モデルに統合してパフォーマンスを向上させることができるだろうか?

私たちのアプローチは非常にシンプルです。Foldseek を使用してタンパク質の構造情報を座標形式から個別のトークンに変換し、それによってアミノ酸語彙と構造語彙を構築し、次にこれら 2 つの語彙を組み合わせて新しい語彙、つまり構造認識語彙 (SA トークン) を生成します。このようにして、元のアミノ酸配列を新しいアミノ酸配列に変換できます。この配列では、大文字はアミノ酸トークンを表し、小文字は構造トークンを表します。その後、マスク言語モデルの作業を継続できます。これを基に、64 個の A100 GPU を使用して 6 億 5000 万パラメータの SaProt モデルをトレーニングしました。トレーニングの総時間はおよそ 3 か月でした。
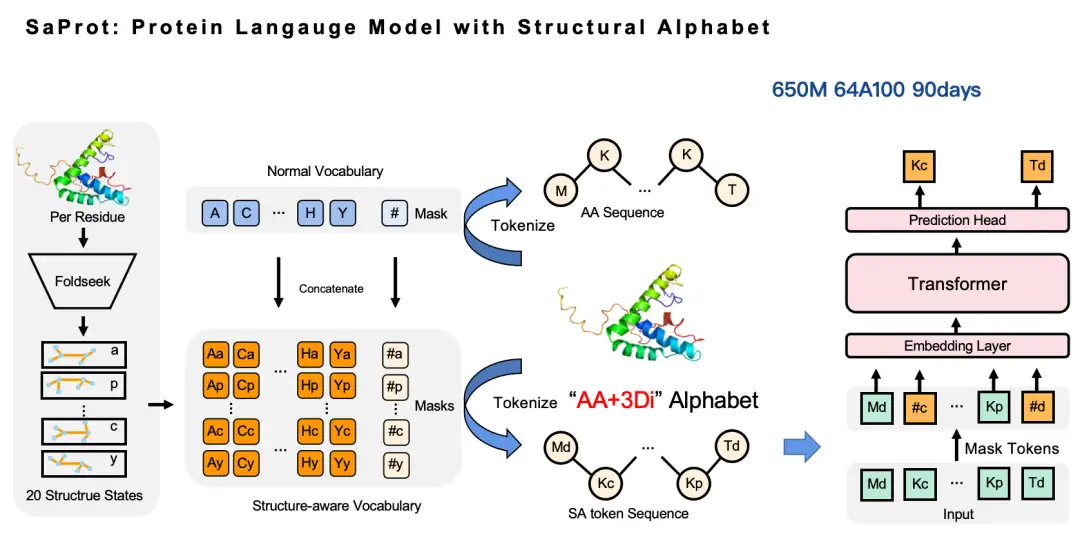
タンパク質構造トークンの変換に Foldseek を選んだのはなぜですか?
Foldseek 3Di トークンのシーケンスを最終的に決定するまでに半年かかりました。直感的には、タンパク質言語モデルに構造情報を組み込むとパフォーマンスが向上するはずですが、実際に試してみると、さまざまな方法を試しましたが失敗しました。たとえば、GNN メソッドを使用してタンパク質構造をモデル化しました。タンパク質構造は実際にはグラフニューラルネットワークであるため、当然タンパク質構造をグラフとしてモデル化したいと考え、MIF 方式を採用しましたが、トレーニングされたモデルの一般化能力が低く、実際の PDB 構造に拡張できないことがわかりました。詳細な分析の結果、マスク言語モデルを使用したモデリング方法が情報漏洩の問題を引き起こすためであると考えられます。
簡単に言えば、AlphaFold によって予測されたタンパク質構造自体には、特定のバイアス、パターン、および AI 予測の痕跡があります。このデータを使用して言語モデルをトレーニングすると、モデルはこれらのトレースを簡単にキャプチャできるため、モデルはトレーニング データに対して優れたパフォーマンスを発揮しますが、一般化能力は低くなります。
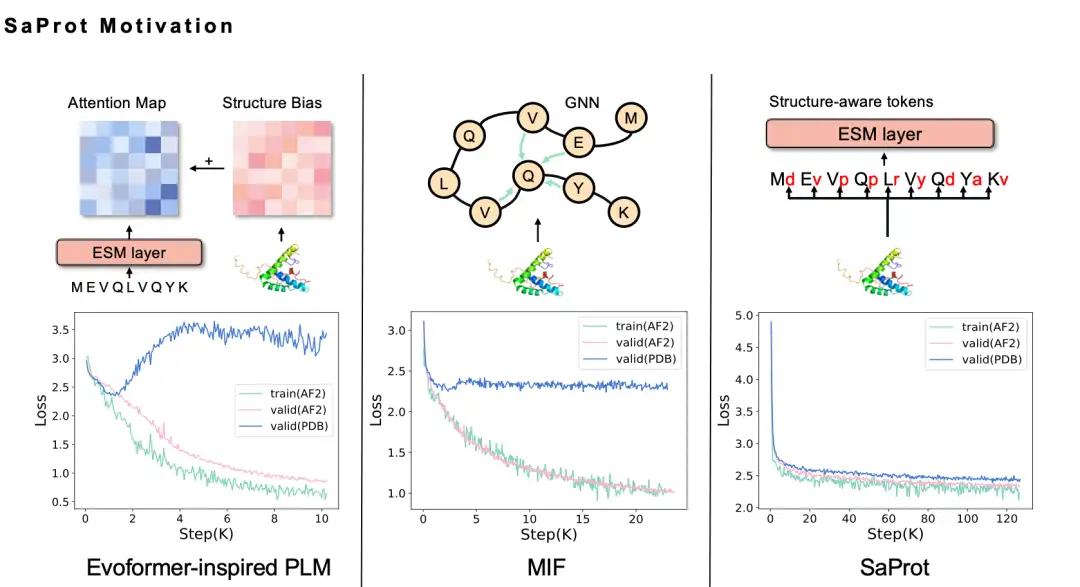
Evoformer方式の使用を含め、さまざまな改善を試みましたが、Foldseekを試すまで情報漏洩の問題は依然として存在していました。 AlphaFold によって予測された構造データで得られた SaProt モデルの損失が削減され、実際の PDB 構造データでの損失も大幅に削減され、期待どおりであることが分かりました。
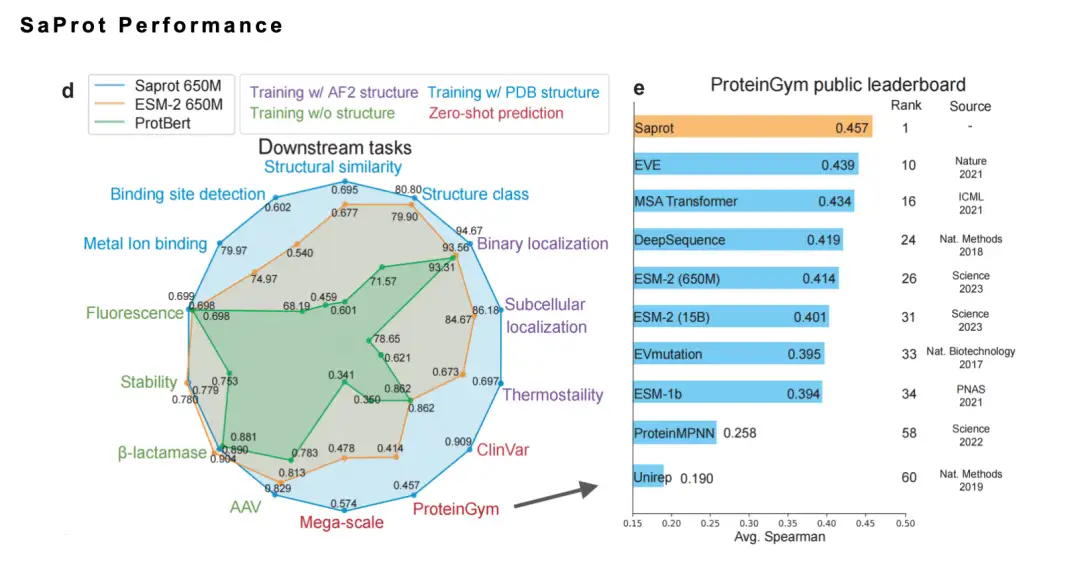
さらに、SaProt は複数のベンチマークで優れたパフォーマンスを発揮します。昨年は権威あるリストProteinGymでも1位を獲得しました。同時に、10種類以上のタンパク質(各種酵素変異修正、蛍光タンパク質修正、蛍光予測など)に対するSaProt/ColabSaProtのコミュニティウェット実験検証結果も収集しましたが、いずれも優れた性能を示しました。
SaProtモデルはかなり良いと思いますが、しかし、多くの生物学者がディープラーニングの訓練を受けていないことを考えると、約10億個のパラメータを持つタンパク質言語モデルを独自に微調整するのは非常に困難です。そこで私たちは、インタラクティブなインターフェース プラットフォーム ColabSaprot + SaprotHub を構築しました。
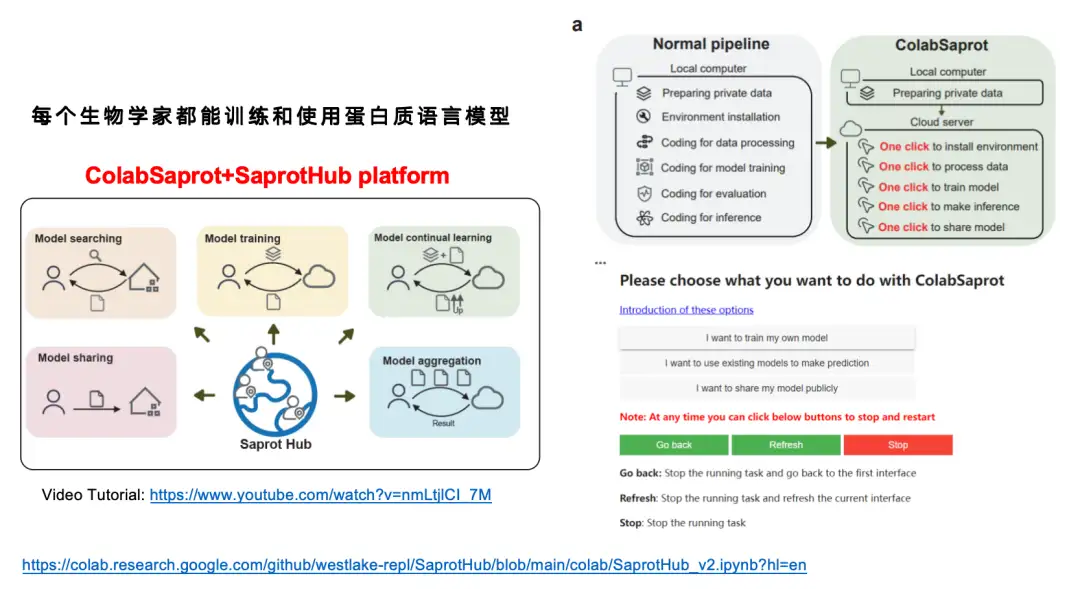
従来のモデルトレーニングプロセス(通常のパイプライン)では、ユーザーはデータの準備、環境の設定、コードの記述、データ処理、モデルのトレーニング、モデルの評価、モデルの推論など、複数のステップを踏む必要があります。ColabSaprot を使用すると、プロセス全体が大幅に簡素化され、ユーザーはいくつかのボタンをクリックするだけで環境のインストール、モデルのトレーニング、予測などの操作を完了できるため、使用のハードルが大幅に下がります。
下の図に示すように、ColabSaprot は主にトレーニング モジュール、予測モジュール、共有モジュールの 3 つの部分で構成されています。

* トレーニング モジュールでは、ユーザーは左側のタスクを記述し、データをアップロードして、トレーニングをクリックするだけです。システムは最適なハイパーパラメータ(バッチ サイズなど)を自動的に選択します。
* 予測モジュールでは、ユーザーは以前にトレーニングしたモデルを直接読み込んで予測を行うことができます。他の研究者が共有したモデルを直接入力して予測を行うこともできます。
* 共有モジュールは、結果を共有しながらデータのプライバシーを保護する方法を提供します。多くの研究室から得られるデータは非常に貴重であり、研究者の中にはこれらのデータをフォローアップ研究に使用する必要がある人もいるかもしれませんが、それでも既存のモデルを共有したいと考えています。 ColabSaprot では、ユーザーはモデル自体のみを共有できます。モデルは本質的にブラックボックスであるため、他の人は元のデータを取得できません。
モデルを共有する場合、言語モデルは通常サイズが大きいため、10億のパラメータを持つモデルをオンラインで直接共有することはほぼ不可能です。そのため、成熟したアダプタ メカニズムを採用しました。ユーザーが共有する必要があるのは、ごく少数のパラメータ(通常は 1% または元のモデルのパラメータの 1/1,000)だけです。誰もがアダプターを互いに共有し、他の人のアダプターを読み込んで、それに基づいて微調整や予測を行うことができます。改善が良好であれば、新しいアダプターを再度共有できるため、効率的なコミュニティ協力メカニズムが形成され、研究効率が大幅に向上します。
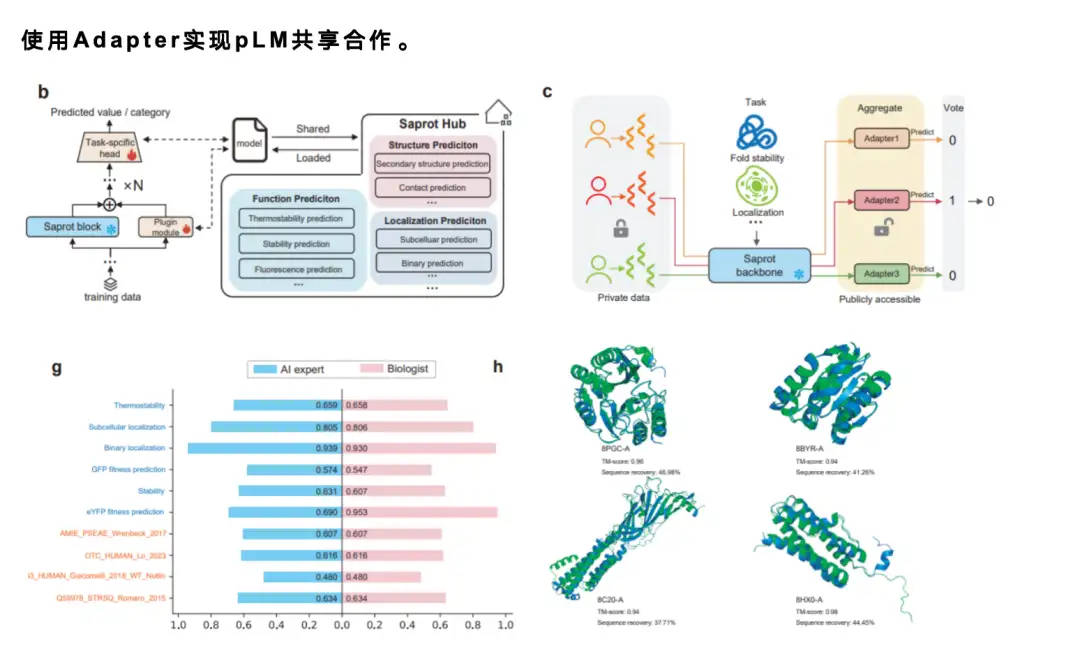
さらに、ユーザー調査も実施しました。機械学習の経験もプログラミングの知識もない 12 人の学生を招待し、ColabSaprot プラットフォームを試用してもらいました。私たちはデータを提供し、実行すべきタスクを伝え、モデルのトレーニングと予測に ColabSaprot を使用するように要求しました。最後に、その結果を AI エキスパートのパフォーマンスと比較すると、これらの非エキスパート ユーザーが ColabSaprot を使用することでエキスパートに近いレベルに到達できることがわかりました。
さらに、タンパク質言語モデルの共有を促進するために、私たちはOPMCというコミュニティも立ち上げました。この分野で国内外の著名な学者が参加し、全員がモデルを共有し、協力とコミュニケーションを促進することを奨励しました。
OPMCアドレス:

ProTrekモデル: タンパク質の配列、構造、機能の対応関係を見つける
2つ目にご紹介したいのは、タンパク質言語モデルProTrekです。
生物学研究では、多くの科学者が次のような問題に直面しています。多くのタンパク質を含むゲノムを持っているものの、その具体的な機能がわからないのです。
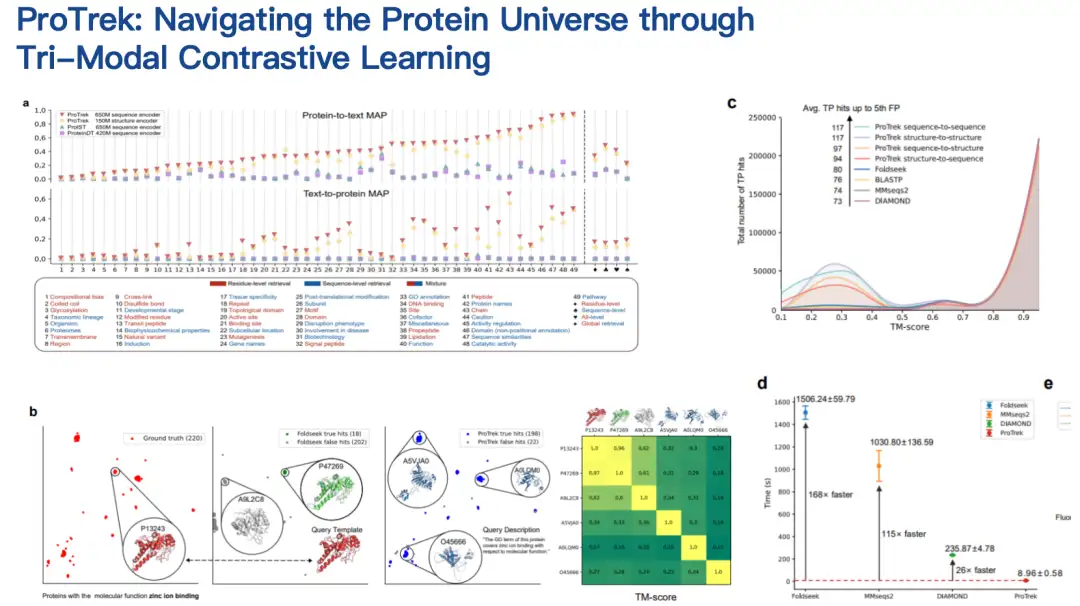
ProTrek は、順序、構造、機能の対照学習のための三様言語モデルです。自然言語検索インターフェースにより、ユーザーは数秒で広大なタンパク質空間を探索し、9 つの異なるタスクについて、配列、構造、機能のすべてのペアの組み合わせ間の関係を検索できます。つまり、ProTrek を使用すると、ユーザーはタンパク質の配列を入力してボタンをクリックするだけで、タンパク質の機能と構造に関連する情報をすばやく見つけることができます。同様に、機能に基づいて配列や構造情報を検索したり、構造に基づいて配列や機能情報を検索したりすることもできます。さらに、配列-配列、構造-構造のクラス検索もサポートしています。
ProTrek使用アドレス:
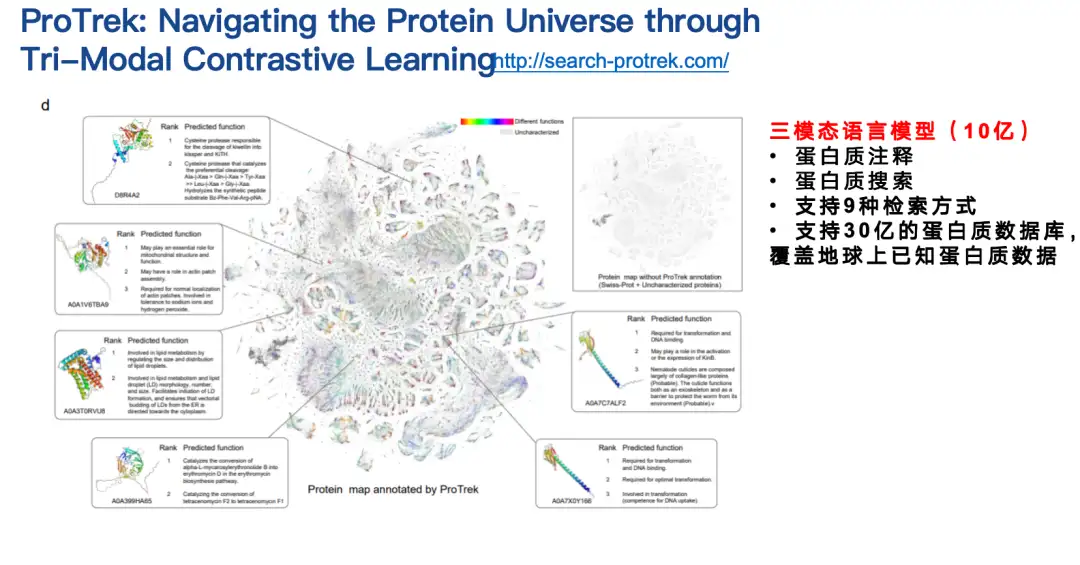
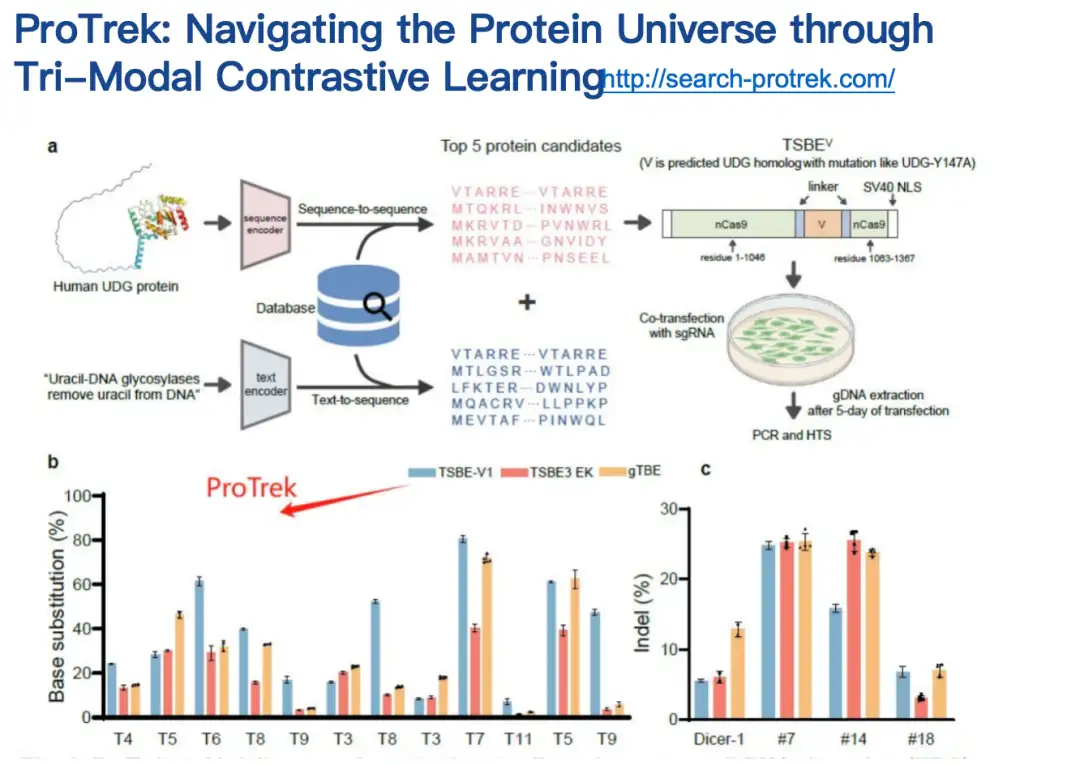
私たちの協力者は、ProTrek モデルをドライテストとウェットテストの両方で評価しました。既存の関連方法と比較して、ProTrek は大幅なパフォーマンスの向上を実現しました。さらに、ProTrek を使用して生成モデルのトレーニング用の大量のデータを生成しましたが、これも優れたパフォーマンスを発揮しました。
Twitterで気づいたのですが多くのユーザーがProTrekを使って競技を始めています。また、多くの肯定的なフィードバックも受け取っており、モデルの実用性がさらに証明されました。
Pinal モデル: テキストを入力するだけで新しいタンパク質配列を設計
私たちのもう一つの研究は、テキスト記述に基づいてタンパク質を設計するためのモデルである Pinal です。
従来のタンパク質設計では通常、生物物理学的エネルギー機能テンプレート情報などの複雑な要素を考慮する必要があります。私たちが探求したいのは、大規模な言語モデルが多くのタスクで優れたパフォーマンスを発揮するので、テキストベースのタンパク質言語モデルを設計できるかどうかです。このモデルでは、タンパク質のアミノ酸配列を設計するために、タンパク質の情報だけを記述すればよいのでしょうか?
Pinal使用アドレス:
http://www.denovo-pinal.com/
用紙のアドレス:
https://www.biorxiv.org/content/10.1101/2024.08.01.606258v1
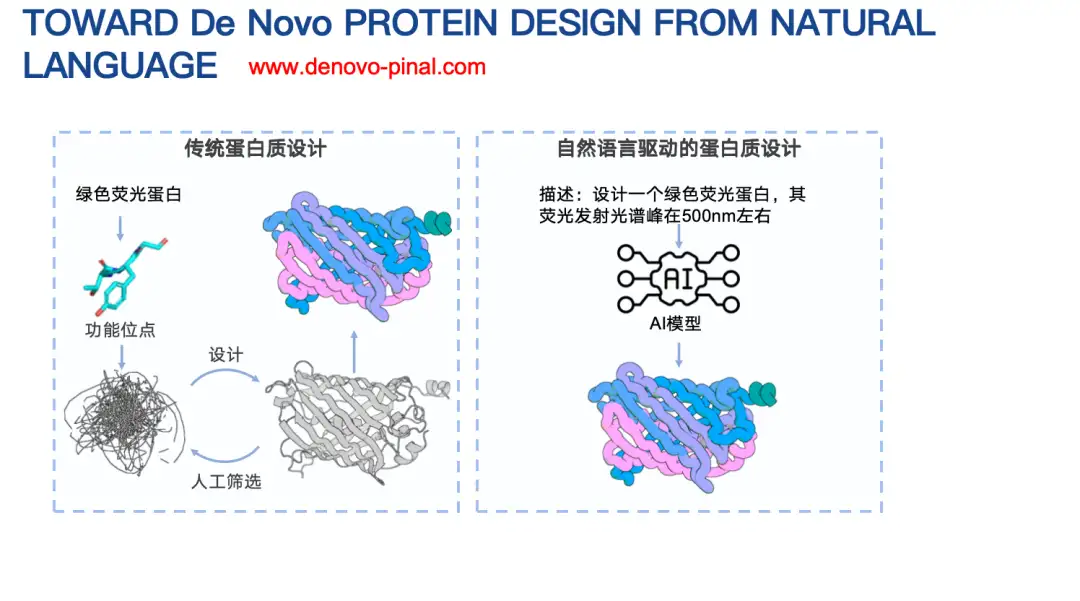
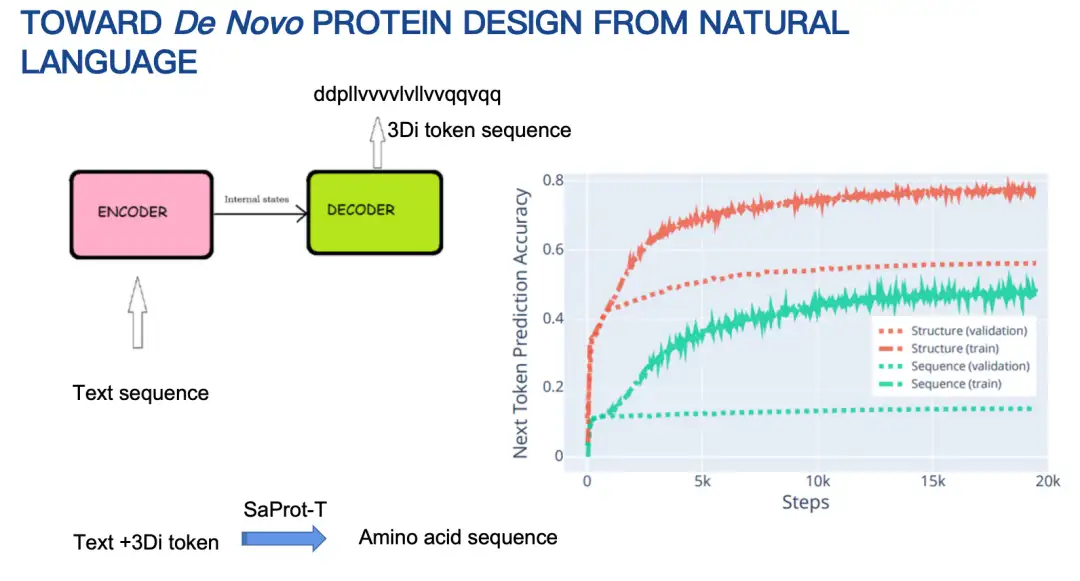
Pinal(160億パラメータ)の基本原理を簡単に紹介します。当初、私たちのアイデアは、テキストを入力してアミノ酸配列を出力するエンコーダー/デコーダー アーキテクチャを使用するというものでした。しかし、長い間試してみたものの、結果は理想的ではありませんでした。主な理由は、アミノ酸配列の空間が広すぎるため、予測が困難になることです。
そのため、私たちは戦略を調整し、まずタンパク質構造を設計し、次にその構造とテキストの手がかりに基づいてアミノ酸配列を設計しました。ここでもタンパク質構造は離散化されたエンコーディングによって表現されます。結果は、下図に示すように、構造と組み合わせた設計方法は、次のトークン予測精度の点で、アミノ酸配列を直接予測する方法よりも大幅に優れていることを示しています。
最近、協力者から Pinal のウェットラボ検証を受け取りました。Pinal は 6 つのタンパク質配列を設計し、そのうち 3 つが発現され、2 つの配列が対応する酵素触媒活性を持つことが検証されました。この研究では、野生型よりも優れたタンパク質の設計を重視しなかったことは言及する価値があります。私たちの主な目標は、テキストに基づいて設計されたタンパク質が対応するタンパク質機能を持っているかどうかを確認することです。
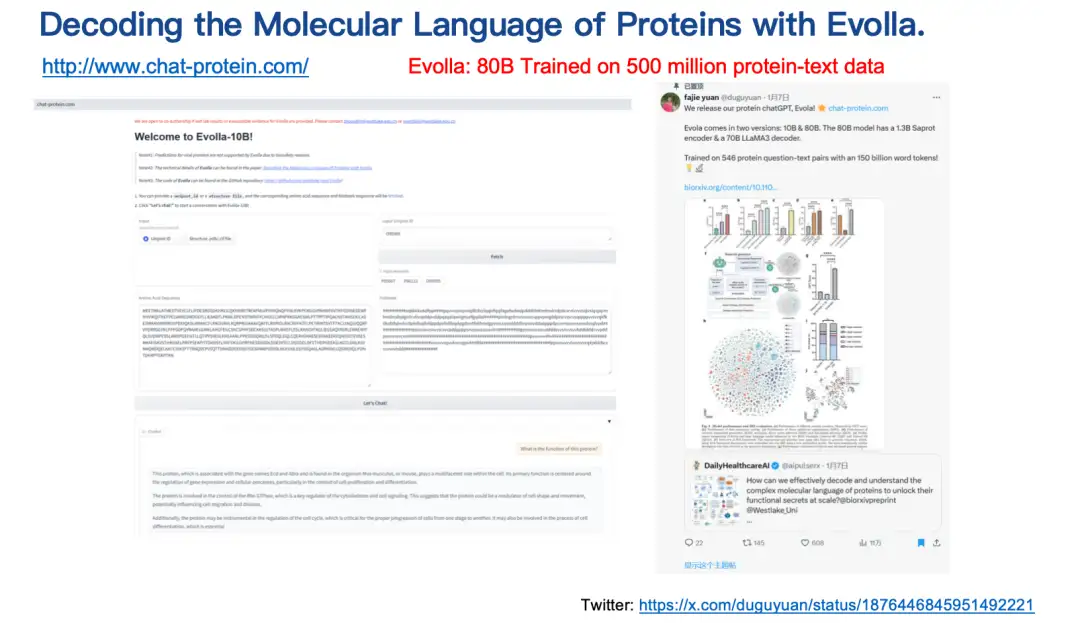
エヴォラモデル: タンパク質の分子言語を解読する
最後に紹介する結果は、Evolla モデルです。これは、タンパク質の分子言語を解読するために設計された、最大規模のオープンソース生物学的モデルの 1 つである、800 億のパラメータを持つタンパク質言語生成モデルです。
タンパク質の配列、構造、ユーザークエリ情報を統合することで、Evolla はタンパク質の機能に関する正確な洞察を生み出します。ユーザーはタンパク質の配列と構造を入力し、タンパク質の基本的な機能や触媒活性の紹介などの質問をしてボタンをクリックするだけで、Evolla が約 200 ~ 500 語の詳細な説明を生成します。
Evollaの使用アドレス:
http://www.chat-protein.com/
Evolla 紙の住所:
https://www.biorxiv.org/content/10.1101/2025.01.05.630192v2

Evolla プロジェクトに必要なトレーニング データと計算能力が膨大であることは言及する価値があります。私たちの博士課程の学生 2 名は、トレーニング データの収集と処理だけでほぼ 1 年を費やしました。最終的に、数千億の単語トークンをカバーする合成データを通じて、5 億を超える高品質なタンパク質テキストのペアを生成しました。このモデルは酵素の機能を非常に正確に予測します。しかし、幻想の問題は避けられません。
チームについて
西湖大学の袁法傑博士は、主に伝統的な機械学習と学際的な科目に関する応用科学研究に従事しており、AIビッグモデルと計算生物学の探求に重点を置いています。彼は、機械学習と人工知能の分野におけるトップクラスの会議やジャーナル(NeurIPS、ICLR、SIGIR、WWW、TPAMI、Molecular Cell など)で 40 本以上の学術論文を発表しています。チームメンバーとプロジェクト貢献者の詳細については、論文を参照してください。
研究グループは機械学習とAI + バイオインフォマティクスの分野で長期的な研究を行っています。研究グループの博士課程学生、研究助手、ポスドク研究員、研究者のポジションに応募することを歓迎します。学生はインターンシップのために研究室を訪問することを歓迎します。ご興味のある方は、[email protected] まで履歴書をお送りください。








