Command Palette
Search for a command to run...
シンガポール国立大学の張楊氏のチームは、複数のベンチマークテストでSOTAを上回る第2世代のRNA構造予測アルゴリズムを開発した。

RNA 分子の構造と機能を理解することは、分子生物学と製薬業界において常に中核的な研究方向となっています。 RNA、特に非コードRNA(ncRNA)は、特定の構造に折り畳まれ、遺伝子調節(転写や翻訳など)、触媒、生物学的シグナル伝達、ストレス反応などのさまざまな細胞プロセスにおいて重要な役割を果たします。
ハイスループットシーケンス技術の急速な発展により、RNA シーケンスデータは飛躍的に増加しましたが、既知のシーケンスと実験的に解明された RNA 構造との間のギャップは広がっています。したがって、RNA の生の配列のみに基づいて RNA の原子構造を解明することがますます緊急になってきています。研究者は、X 線結晶構造解析、核磁気共鳴分光法、極低温電子顕微鏡 (cryo-EM) などの構造生物学技術など、RNA 構造を研究するためのさまざまな方法を開発してきました。これらの実験技術はより高い解像度を提供できますが、RNA の 3 次元構造の実験的解明は多くの場合コストがかかり、場合によっては達成が困難です。したがって、配列から直接、高品質の RNA の 3 次元構造を予測する計算方法に対する需要が高まっています。
「アブイニシオRNA構造予測」とは、実験データや事前の知識に頼らずに、RNAの配列からRNAの3次元構造を直接予測する方法を指します。この方法の核心は、コンピューターシミュレーションと計算化学技術を使用して、数学モデルとアルゴリズムを通じて RNA 分子の 3 次元構造を予測することです。
最近、シンガポール国立大学の張楊教授チームの最新の研究成果により、「第一原理RNA構造予測」がさらに高いレベルにまで進歩しました。研究者らは、ディープラーニングに基づく高精度のRNA構造予測フレームワーク、DRfold2を提案した。事前トレーニング済みの RNA 複合言語モデル (RCLM) とノイズ除去構造モジュールを統合し、エンドツーエンドの RNA 構造予測を実現します。 DRfold2 は、複数のベンチマークにおいて他の最先端の方法と比較して、グローバル トポロジーと二次構造予測の両方で優れたパフォーマンスを発揮します。
詳細な分析により、この改善は主に、RCLM の共進化パターンを捕捉する能力と効率的なノイズ除去プロセスによるものであることが示されています。これにより、既存の方法と比較して、DRfold2 の教師なし接触予測精度が 100% 以上向上します。
関連する結果は、「複合言語モデルとノイズ除去エンドツーエンド学習によるアブイニシオRNA構造予測」というタイトルでプレプリントプラットフォームbioRxivに掲載されています。
研究のハイライト:
* DRfold2は、事前にトレーニングされたRNA複合言語モデル(RCLM)とノイズ除去構造モジュールを統合し、エンドツーエンドのRNA構造予測を実現します。
* 複合言語モデリング、ノイズ除去ベースのエンドツーエンド学習、ディープラーニングによるポスト最適化のユニークな組み合わせにより、DRfold2 は「Ab initio RNA 構造予測」の新しい方向性を切り開きます。
* DRfold2はAlphaFold3と非常に補完的であり、最適化フレームワークに統合すると統計的に有意な精度の向上を実現します。

用紙のアドレス:
https://www.biorxiv.org/content/10.1101/2025.03.05.641632v1
DRfold2 RNA構造テストデータセットをダウンロード:
データセット: 独立したテストデータセットを構築する
DRfold2の性能を客観的に評価するために、研究者らは、28 個の RNA 構造を含む独立したテスト データセットを構築しました。それらの配列の長さはすべて 400 nt 未満であり、次の 3 つのカテゴリに分類されます。
* 最新のRNAパズルのターゲット配列
* CASP15競争におけるRNA標的配列
* 2024年8月1日現在、タンパク質データバンク(PDB)データベースに最近公開されたRNA構造
注目すべきことに、研究者らは、機能解析と医薬品設計の主な焦点である自然界で発見されたRNA構造から逸脱しているという理由で、大規模な合成RNA構造をCASP15データセットから除外した。
厳密なモデル評価を確実に行うために、トレーニング セットには 2024 年以前に公開された RNA 構造のみが含まれ、テスト データセットとの配列類似性が 80% を超える RNA は除外されます。
DRfold2 RNA構造テストデータセットをダウンロード:
モデルアーキテクチャ: 新しい RNA 3D 構造予測パイプライン DRfold2
DRfold2 は、新しい RNA 3D 構造予測パイプラインで、次の 4 つのコア モジュールで構成されています: (1) RNA 複合言語モデル (RCLM)、(2) RNA トランスフォーマー ブロック、(3) ノイズ除去構造モジュール、(4) CSOR プロトコルによる最終モデルの選択と最適化 (下の図 A を参照)。
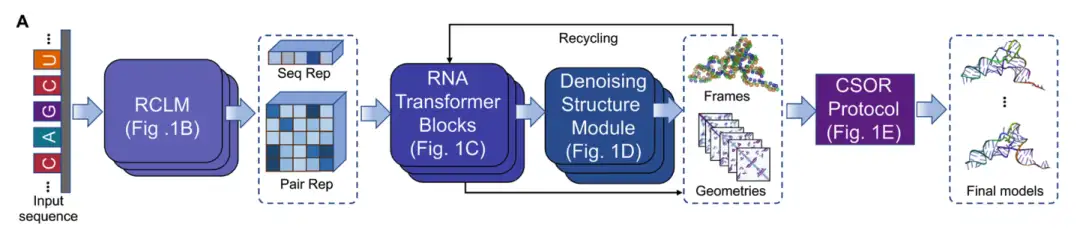
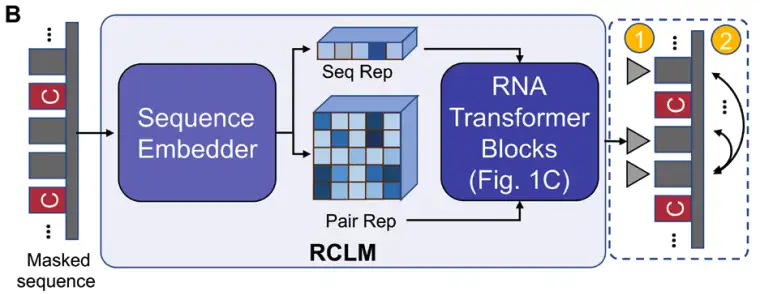
入力RNA配列から始めて、DRfold2 はまず、事前トレーニング済みの RNA 複合言語モデル (RCLM) を使用してクエリ シーケンスをエンコードします。シーケンス表現 (Seq Rep) とペア表現 (Pair Rep) を生成します。 RCLM は、複合尤度最大化法を通じて大規模な教師なしシーケンス データでトレーニングされ、下の図 B に示すように、より効率的なシーケンス パターン認識を実現します。
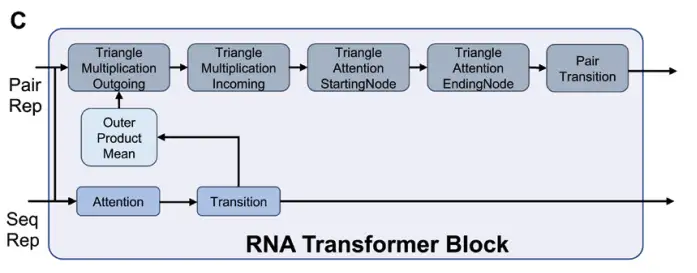
これらの配列とペアワイズ表現は、RNA Transformer モジュールに送られ、RNA 構造の折り畳みに必要な主要な特徴表現を生成する処理が行われます (下の図 C を参照)。
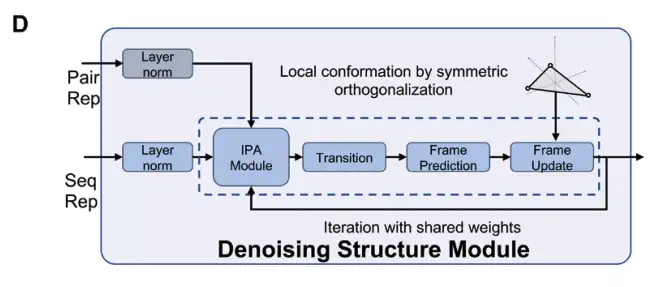
次に、DRfold2 は、デノイズ RNA 構造モジュール (DRSM) を使用して、下の図 D に示すように、エンドツーエンド方式で RNA コンフォメーションを生成します。
最終的な RNA 構造モデルは、後処理 CSOR プロトコルを通じてスクリーニングおよび最適化され、複数のチェックポイントで生成されたコンフォメーションのセットから最適なモデルが選択され、改良されます (下の図 E を参照)。
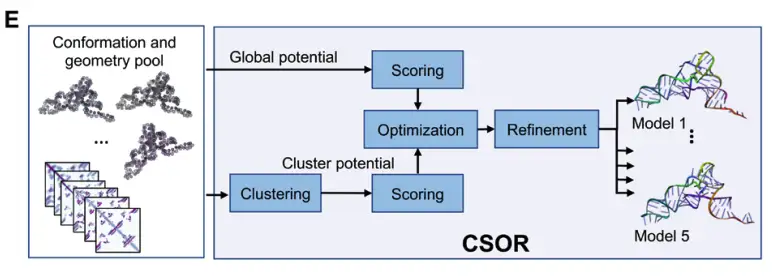
DRfold2 は、チームの以前の DRfold メソッドと似た名前が付けられていますが、まったく異なるフレームワークに基づく大きな進歩を導入しています。最も重要なのは、複合言語モデルの統合であり、これにより RNA シーケンスとペアの表現能力が大幅に向上します。さらに、予測パイプラインにはノイズ除去 RNA 構造モジュール (DRSM) が統合されており、制御された摂動戦略を使用して、ノイズの多い RNA 構造を効率的に修正することで構造変換を堅牢に学習します。
研究者らは、DRfold2 オンライン サーバーとローカル コードを次の場所で公開しています。
https://zhanglab.comp.nus.edu.sg/DRfold2
研究結果: DRfold2 は複数のベンチマークで他の最先端の方法よりも優れている
研究者らはまず、RNAComposer(フラグメントアセンブリと最適化ベース)、trRosettaRNA(ディープラーニング法)、RhoFold(エンドツーエンドディープラーニング法)、RoseTTAFoldNA(エンドツーエンドディープラーニング法)、DeepFoldRNA(ディープラーニング法)を含む5つの最先端のRNA構造予測法とDRfold2を比較しました。
下の図に示すように、研究者らは、異なる配列類似性閾値(50%-80%)における DRfold2 とベンチマーク法の TM スコアと RMSD 評価結果を比較しました。その中で、TM スコアは、予測された RNA 構造の全体的な品質を評価するために使用される長さに依存しないスコアリング関数です。値の範囲は0〜1です。値が高いほど、予測された構造と実際の構造の類似性が高くなります。
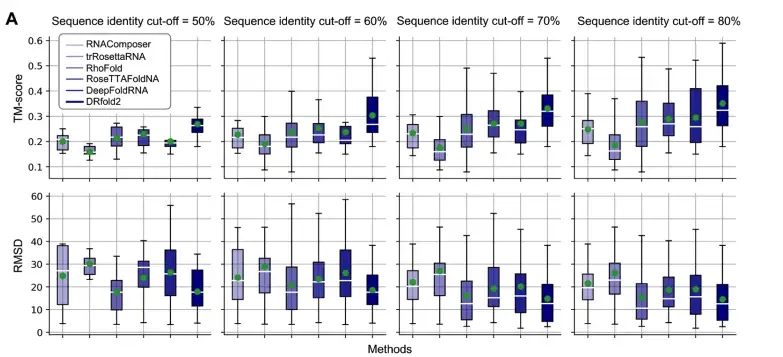
結果は、DRfold2 がすべての配列類似性しきい値の下で常に最高の平均 TM スコアを取得することを示しています。例えば:
* 80% 類似性しきい値では、DRfold2 の平均 TM スコアは 0.351 であり、2 位の DeepFoldRNA (TM スコア = 0.296) よりも 18.6% 高くなります。
* 50% 類似性しきい値 (最も厳しいテスト セット) の下でも、DRfold2 は平均 TM スコア 0.269 を取得できます。これは、2 位の RoseTTAFoldNA (TM スコア = 0.229) よりも 17.5% 高い値です。
* さらに、すべての配列類似性しきい値における DRfold2 の RMSD (二乗平均平方根偏差) は、すべてのコントロール方法よりも常に低く、予測された構造が実際の RNA 構造に近いことを示しています。
研究者らはさらに、チンパンジーの CPEB3 HDV 様リボザイム (PDB ID: 7QR3) を例として使用しました。 RNA は 69 ヌクレオチドの長さがあり、さまざまな方法による RNA 三次構造の予測効果を分析しました。結果は次のとおりです。
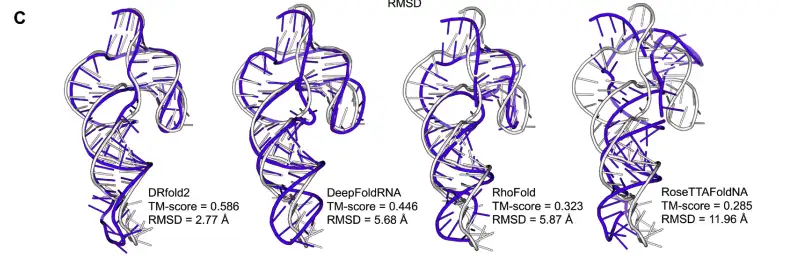
* DRfold2 は、TM スコアが 0.586、RMSD がわずか 2.77 Å で、リボザイムの全体的なトポロジー構造を正確に捉えました。
* DeepFoldRNA は全体的ならせん配置に関しては優れたパフォーマンスを発揮しますが、ヘアピン ループの方向が大幅にずれているため、RMSD は 5.68 Å にも達し、DRfold2 の偏差の 2 倍になります。
* RhoFold と RoseTTAFoldNA はジャンクション領域での空間予測の誤差が大きく、TM スコアがそれぞれ 0.323 と 0.285 に低下します。
* ターゲット RNA とトレーニング データセット間の最高の配列類似性は 60.9% のみであり、これは DRfold2 が相同テンプレートが存在しない場合でも新しい RNA 配列に対して信頼性の高い構造予測を提供できることを示しています。
これらの結果は次のことを示しています:RCLM のような高次言語モデルによって提供される包括的な確率的表現は、共進化パターンと空間的制約を学習する能力を大幅に強化します。このように、DRfold2 のエンドツーエンド ネットワークを通じて、より正確な 3D RNA 構造モデリングが実現しました。
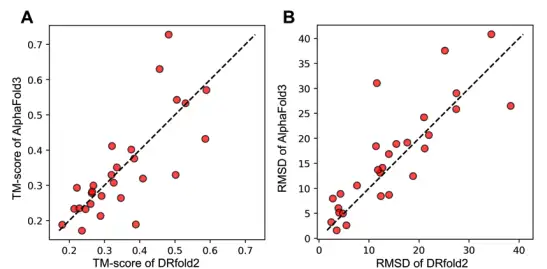
これを基に、RNA 3D構造予測におけるDRfold2とAlphaFold3のパフォーマンスを比較するために、研究者らはテストセット内のRNA配列をAlphaFoldサーバーに送信し、デフォルトのシード構成を使用してAlphaFold3の予測構造を取得しました。消す、DRfold2 の平均 TM スコア (0.351) と RMSD (14.6 Å) は、AlphaFold3 (0.345 と 16.0 Å) よりもわずかに高くなっています。
さらに注目すべきは、DRfold2 と AlphaFold3 は全体的に同様のパフォーマンスを示していますが、下の図の結果は、特に予測が対角線から大きく外れている場合に、両者の強い補完性を強調しているということです。AlphaFold3 の予測を DRfold2 最適化フレームワークに追加の潜在的な機能項として組み込むことで、研究者は TM スコアと RMSD の両方で統計的に有意な改善を達成しました。
張楊教授のチームは長年AIと計算生物学の研究に注力してきた。
この研究で提案された DRfold2 は、実際には、以前に張楊教授のチームによって提案された DRfold モデルのアップグレード版です。
2023年9月、張楊教授のチームは「エンドツーエンド学習と深層幾何学的ポテンシャルの統合による第一原理RNA構造予測」と題する論文をNature Communications誌に発表した。
この研究では、RNA の 3 次元構造を正確に予測する新しい技術 DRfold について報告します。中核となる革新は、FAPE ポテンシャルと幾何学的ポテンシャルという 2 つの補完的なポテンシャルエネルギー関数の導入にあります。これらは 2 つの独立した Transformer ネットワークを介してトレーニングされ、一緒に RNA 構造予測のディープラーニングの可能性を構成します。計算結果によると、以前の RNA 構造コンピュータ予測方法と比較して、DRfold は多くのパフォーマンス指標でこれらの方法を上回っています。

用紙のアドレス:
https://www.nature.com/articles/s41467-023-41303-9
DRfold から DRfold2 まで、張楊教授のチームは長年にわたり人工知能と計算生物学の研究に注力し続けてきました。彼の研究室は、ディープラーニングに基づいたタンパク質やRNAの構造予測研究を最も早くから行っている研究室の一つです。米国スローン賞、米国国立科学財団キャリア賞、ミシガン大学基礎科学研究賞などの栄誉を獲得しています。 2015年以降、トムソン・ロイター/クラリベイト・アナリティクスの「Global Highly Cited Scientists」リストに7回選出されています。彼の研究室で開発されたI-TASSERアルゴリズム(https://zhanggroup.org/I-TASSER/), 2006年以来、世界中のCASP実験において9回連続で最も正確な自動タンパク質構造予測法として評価されています。
2024年1月2日、張楊教授のチームは「膨大なメタゲノムデータを用いたDeepMSA2を用いたディープラーニングによるタンパク質モノマーと複合体構造予測の改善」と題する論文をNature Methods誌に発表した。
この研究では、タンパク質相互作用の構造予測の精度を向上させるために 2 つの新しいソフトウェアが開発されました。著者らは、再帰的動的プログラミングと隠れマルコフモデルアルゴリズムを使用して、大規模なメタゲノム配列ライブラリから高品質の MSA データを迅速に抽出し、新しく開発された DMFold ソフトウェアを使用してタンパク質複合体の 3 次元構造を構築する DeepMSA2 を開発しました。
実験結果によると、タンパク質複合体に対する DMFold/DeepMSA2 の構造予測精度は、AlphaFold2 などのアルゴリズムよりも大幅に優れています。特に、DMFold(https://zhanggroup.org/DMFold) アルゴリズムが最新のタンパク質構造予測コンペティション (CASP15) においてタンパク質複合体構造予測部門で優勝しました。

用紙のアドレス:
https://www.nature.com/articles/s41592-023-02130-4
最近、チームは研究の方向性をさらに拡大し、RNA と短いペプチドの設計と構造予測を含め、薬物設計に関連するトピックを探求しました。今後も張楊教授はチームを率いて生物学の謎の探求を続けていくと信じています。
参考文献:
1.https://www.biorxiv.org/content/10.1101/2025.03.05.641632v1
2.https://mp.weixin.qq.com/s/X_VJ-WOWEP08p5GAJOgq9A








