Command Palette
Search for a command to run...
AlphaFold がアプリケーションにおいて新たなマイルストーンに到達しました!ケンブリッジ大学のチームは、無秩序なタンパク質構造セットを正確に予測するためのAlphaFold-Metainferenceを提案した。
2018年末のAlphaFoldの登場以来、タンパク質構造予測の分野はAIのサポートにより大きな変化を遂げてきました。現在、AlphaFold は予測精度で優れた性能を発揮しているだけでなく、最新のバージョンでは予測範囲も徐々に拡大しています。中国科学院の士一公院士がメディアの前で「私の意見では、これは人工知能が科学分野にもたらした最大の貢献である。また、21世紀に人類が成し遂げた最も重要な科学的進歩の一つでもある。人類による自然界の科学的探究において、非常に注目すべき歴史的成果である」とコメントしたのも不思議ではない。
AlphaFold が主導するタンパク質構造予測革命は非常に急速ですが、まだ解決されていない問題がいくつか残っています。中でも、無秩序なタンパク質の研究は生命科学の分野において常に難しい問題となってきました。これらのタンパク質は、細胞シグナル伝達、制御プロセス、およびさまざまな疾患において重要な役割を果たします。しかし、構造における特殊な異質性とダイナミクスのため、単一の構造で表現することはできません。そのため、その研究は、秩序だったタンパク質構造の予測ほど大きな進歩を遂げていません。 AlphaFold の成功は、科学者にとって問題を解決する新たな方法を示しました。
最近、ケンブリッジ大学の研究チームが新たな研究を発表し、AlphaFold-Metainferenceと呼ばれる手法を提案しました。この方法では、AlphaFold によって予測された予測整列誤差 (PAE) マップと分子動力学 (MD) シミュレーションの距離変化マトリックス間の相関関係を利用して、無秩序なタンパク質と無秩序な領域を含むタンパク質の構造アンサンブルを構築します。これは、ディープラーニング手法に基づく無秩序なタンパク質構造の予測に新しいアイデアを提供し、AlphaFold の適用範囲をさらに広げます。
現在、当該研究成果は「無秩序タンパク質の構造アンサンブルのAlphaFold予測」というタイトルで国際学術誌Nature Communicationsに掲載されています。
研究のハイライト:
* 予測の限界を打ち破り、高精度な予測を実現します。この研究では、AlphaFold は無秩序なタンパク質データでトレーニングしなくても残基間の距離を正確に予測できることが確認されました。
* 予測方法を革新し、構造コレクションを構築します。この方法では、AlphaFold によって予測された距離を構造制約として使用し、メタ推論フレームワークと分子動力学シミュレーションを組み合わせて、無秩序なタンパク質と無秩序な領域を含むタンパク質の構造のコレクションを構築します。
* ディープラーニング手法を深め、アプリケーションの境界を拡大します。この方法は、高度に無秩序なタンパク質や部分的に無秩序なタンパク質の処理に効果的です。生成された構造コレクションは、単一の AlphaFold 構造よりも実験データとの一貫性が大幅に高く、無秩序なタンパク質構造の予測の問題を効果的に解決します。

用紙のアドレス:
https://www.nature.com/articles/s41467-025-56572-9
オープンソース プロジェクト「awesome-ai4s」は、200 を超える AI4S 論文の解釈をまとめ、膨大なデータ セットとツールを提供します。
https://github.com/hyperai/awesome-ai4s
データセット: 複数のソースデータの厳密な検証
ディープラーニングモデルのトレーニングに関しては、無秩序なタンパク質の構造コレクションは数も精度も非常に低いものの、秩序だったタンパク質の利用可能な情報に基づいて無秩序なタンパク質を予測できるため、研究者はタンパク質データバンク(PDB)内の多数の高解像度の折り畳まれたタンパク質構造を使用してディープラーニングモデルをトレーニングしました。
実験データの比較に関しては、無秩序なタンパク質の残基間距離に関する実験情報を取得することは困難であり、データラベル自体がコンフォメーションアンサンブルの特性に影響を及ぼす可能性があります。これを実行するために、研究者らは小角X線散乱(SAXS)データと核磁気共鳴(NMR)拡散測定を使用しました。研究用に無秩序なタンパク質残基間の距離分布に関するラベルフリー情報を提供し、予測結果の比較と検証に使用されます。
さらに検証してみると、研究者らはまた、CALVADOS-2(C2)を用いた全原子分子動力学シミュレーションと粗視化シミュレーションを通じて得られたAβとαシヌクレインの構造アンサンブルデータを分析した。これにより、AlphaFold の予測距離の精度がさらに検証されます。
モデルアーキテクチャ: 革新的な融合メタ推論法
この研究で説明した AlphaFold-Metainference 法は、無秩序なタンパク質および無秩序な領域を含むタンパク質の天然の状態を表す構造のコレクションを生成するために使用されます。
このアプローチの核心は、AlphaFold によって予測される残基間の距離が無秩序なタンパク質の場合でも比較的正確であり、したがってメタ推論フレームワーク内の分子動力学シミュレーションにおける構造的制約として使用できるという観察に基づいています。簡単に言えば、構造アンサンブルを生成するために、AlphaFold-Metainference は予測距離を分子動力学シミュレーションにおける構造制約として使用します。AlphaFold 距離マップ (ディストグラム) を構造セットに変換します。
まず、AlphaFold 予測距離です。研究者らは、AlphaFold の距離マップを使用して残基間の平均距離を予測し、特定の式を使用して予測距離と標準偏差を計算しました。次に、MMseqs2に基づいて多重配列アライメントを行い、構造テンプレートを使用せずにデフォルト設定のAlphaFold 1.1.1モデルを使用して予測を行いました。 AlphaFold によって出力される残基間距離は、2.15625 Å から 21.84375 Å の範囲の等幅の 64 個のビンに分散され、最後のビンには 21.84375 Å を超える距離も含まれます。
次に、メタ推論法を組み合わせます。いわゆるメタ推論はベイズ推論法であり、最大エントロピー原理に基づいて事前情報と実験データを組み合わせて構造セットを決定することができます。この段階では、研究者らは、AlphaFoldによって予測された距離グラフを疑似実験データとして使用し、ベイズメタ推論法を適用した。力場やフォワード モデルの不正確さ、データ内のランダム エラー、アンサンブル内のサンプル サイズの制限によるエラーなどの体系的なエラーから構造の異質性を分離して、構造アンサンブルを決定します。
分子動力学シミュレーションでは、メタ推論エネルギー関数に基づいて計算が行われ、複数のレプリカシミュレーションとギブスサンプリングを通じて誤差パラメータが決定されます。最後に、CALVADOS-2 力場を使用して粗粒度シミュレーションを実行しました。AlphaFold-Metainference を実装します。
最後のステップは距離制約の選択です。この段階では、距離確率と予測されたアライメント誤差に基づいて、AlphaFold によって予測された距離が選択されます。選択基準は、タンパク質の親水性と予測局所距離差テスト (pLDDT) スコアを組み合わせて決定されました。構造化領域内の残基距離を選択するために pLDDT スコアを実験的に使用しても、構造アンサンブルの生成を最適化するための距離制約としての使用が排除されるわけではないことに注意する価値があります。
すべての分子動力学シミュレーションは、AlphaFold によって予測された構造から開始され、NVT アンサンブルの下で実行されました。シミュレーションごとに 6 つのレプリカが設定され、各レプリカは 100 万ステップ実行され、エネルギー最小化ステップで取得されたさまざまな初期位置からシミュレーションが開始されました。シミュレーションではランジュバン積分器を使用します。時間ステップは 5 fs、摩擦係数は 0.01 ps⁻¹ であり、CALVADOS-2 パラメータと関数形式を備えた Cα ベースのモデルが使用されます。
その中で、高度に無秩序なタンパク質と部分的に無秩序なタンパク質については、PULCHRA を使用して粗視化コレクション内のすべての構造を全原子表現に変換し、その後 GROMACS を使用してエネルギー最小化を行い、より正確な構造を取得しました。
全体として、研究者らが発表した結果は、もともと折り畳まれたタンパク質の本来の状態を予測するために開発されたディープラーニング手法が、無秩序なタンパク質の本来の状態を表す構造の集合を生成するためにどのように使用できるかを示しています。この方法は、ディープラーニングに基づくタンパク質構造予測の範囲を大幅に拡大し、無秩序なタンパク質構造の予測に新たなアイデアを提供します。
実験結果:その合理性を完全に検証
AlphaFoldの予測精度に関して
研究者らは、SAXS と NMR の両方の拡散測定が可能な 11 種類のタンパク質を比較し、AlphaFold によって予測された距離分布と SAXS から得られた距離分布がよく一致することを発見しました。研究者らは、下の図に示すように、折り畳まれたタンパク質も対照として追加しました。
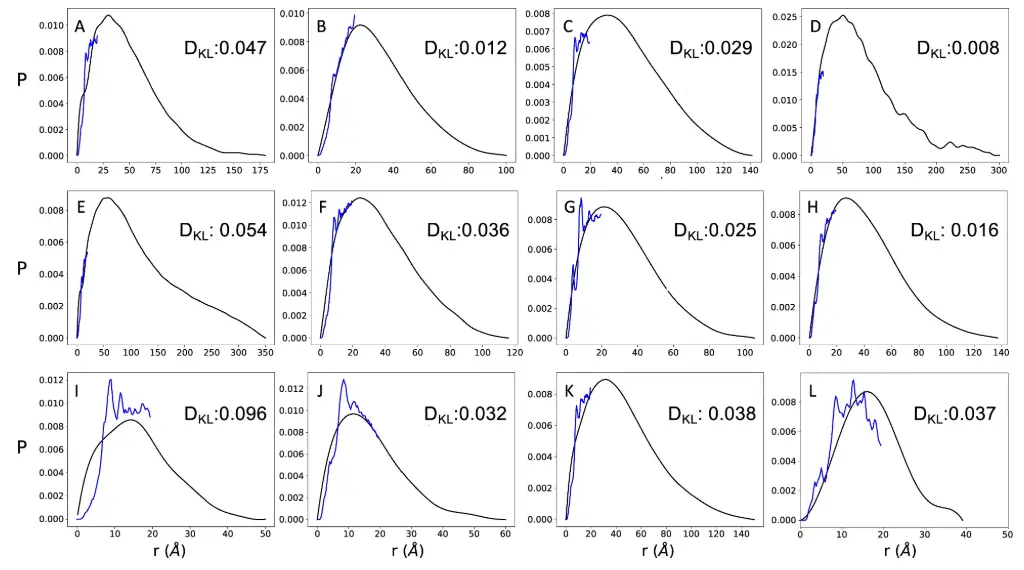
AlphaFold によって予測される距離は最大約 22 Å であるため、AlphaFold によって予測される距離分布は SAXA から導出された分布全体をカバーしていないことに注意してください。結果によると、追加された対照群のDKL値は0.037であり、これは11種類の高度に無秩序なタンパク質のDKL値(DKL範囲は0.008〜0.096)と同等でした。これはさらに、AlphaFold が無秩序なタンパク質と秩序のあるタンパク質の残基間距離を予測する際に同等の精度を持っていることを示しています。
さらに、AlphaFold によって予測された距離は、Aβ および α-シヌクレインの MD アンサンブルおよび CALVADOS-2 アンサンブルから逆計算された距離ともよく一致しています。
高度に無秩序な構造の集合の検証において
ペアワイズ距離分布は、小角X線散乱測定を使用して計算できます。研究者らは、実験的に得られた距離分布と、AlphaFold-Metainference シミュレーションによって決定された構造セットから得られた距離分布を、前述の 11 個の高度に無秩序なタンパク質について再度比較しました。
同時に、研究者らはさらなる比較のために、CALVADOS-2 を使用して得られた距離分布と、単一の AlphaFold 構造から直接生成された AlphaFold 由来の距離分布も示しました。定量的な比較を行うために、研究者らは、CALVADOS-2 を使用した AlphaFold-Metainference によって提供された構造の集合が、単一の AlphaFold 由来の構造よりも SAXS データと一致していることを発見しました。
研究者らはさらに、CamShift を使用して各時間ステップで逆計算された NMR 化学シフトを使用して構造集合体を比較しました。結果は、場合によっては AlphaFold-Metainference の予測の方が正確であることを示しています。以下に示すように。
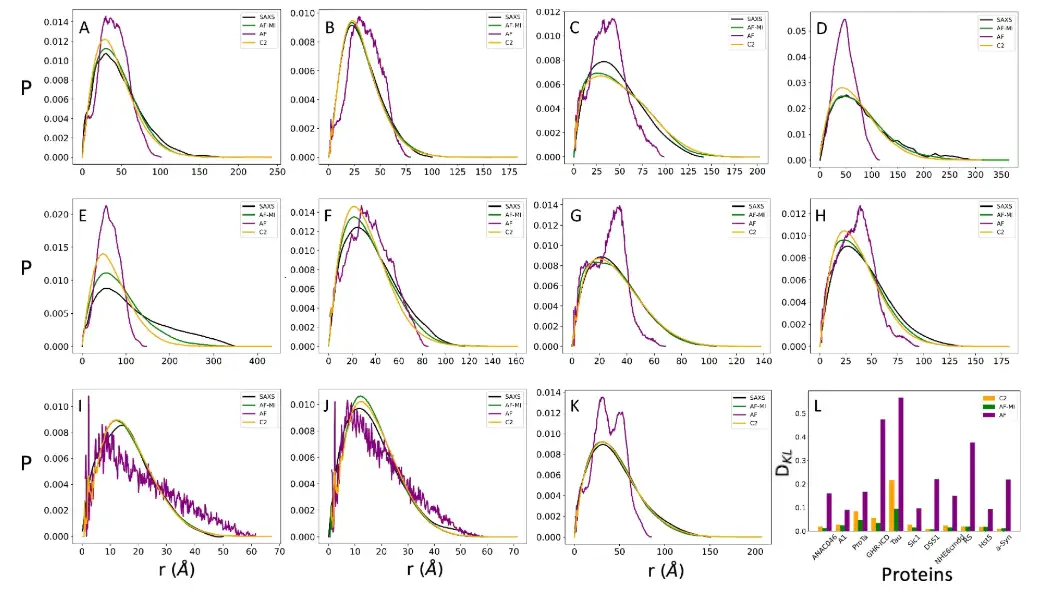
* SAXSで得られた実験的ペアワイズ距離の分布は黒線で示されている。
* AlphaFold単一構造予測は紫色の線で表されます
* AlphaFold-Metainference構造アンサンブル予測は緑の線で表されます
* CALVADOS-2によって得られたペアワイズ距離分布はオレンジ色の線で示されている
部分的に無秩序な構造化コレクションの検証において
研究者らは、配列長が異なり、検証にSAXSデータが利用できる、秩序だったドメインと無秩序なドメインの両方を持つ6つのタンパク質のセットを準備しました。
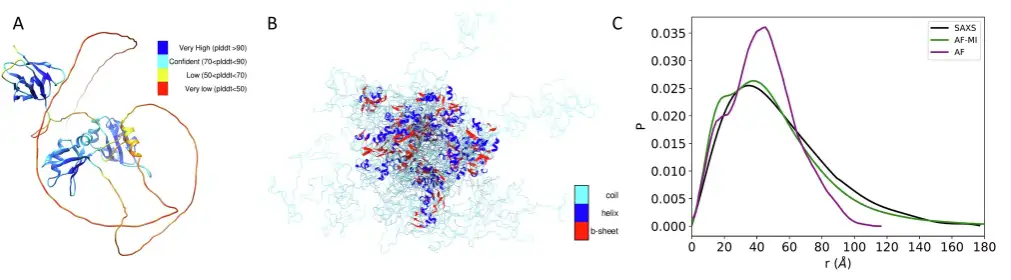
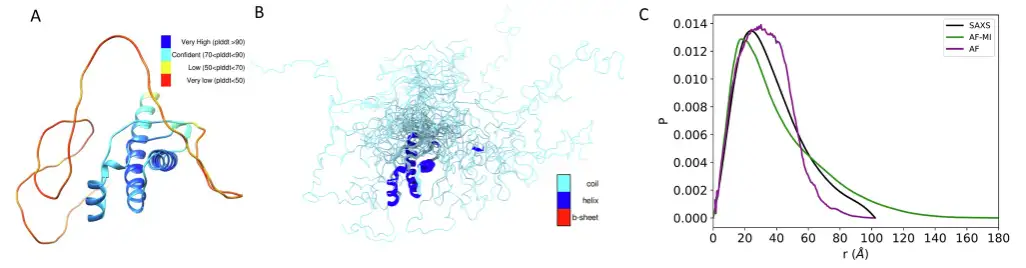
1 つ目は TDP-43 です。これはモジュール構造を持つ多機能 RNA 結合タンパク質で、転写、pre-mRNA スプライシング、mRNA 安定性の調節など、さまざまな細胞プロセスに関与しており、ALS やその他の神経変性疾患に関係していると考えられています。
実験結果によると、研究者のフィルタリング基準を適用してAlphaFoldによって予測された距離を選択し、これらの距離制約を使用してAlphaFold-Metainferenceを適用すると、得られた構造集合は SAXS データと大幅によく一致しています。DKL値はわずか0.018です。これは、AlphaFold 予測構造を SAXS データと直接使用した場合の DKL 値 0.582 よりも優れています。以下に示すように。
研究者らはその後、アタキシン3とヒトプリオンタンパク質の分析を続けた。前者については、上記のTDP-43と同様の結果が得られた。AlphaFoldタンパク質構造データベースから直接AlphaFoldを使用して得られた予測構造は、SAXSデータとの整合性が低く、DKL値は0.653であったが、AlphaFold-MetainferenceシミュレーションからAlphaFold予測距離を選択するためにフィルタリング基準を適用した場合、SAXS データとより一致する一連の構造が得られました。DKL値はわずか0.020です。下の図の通りです。

後者については、AlphaFoldを使用してAlphaFoldタンパク質構造データベースから直接得られた予測構造は、DKL値が0.1であり、SAXSデータとの整合性が低い。フィルタリング基準を適用すると、SAXS データとより一致する構造セットが得られました。DKL値はわずか0.053です。下の図の通りです。
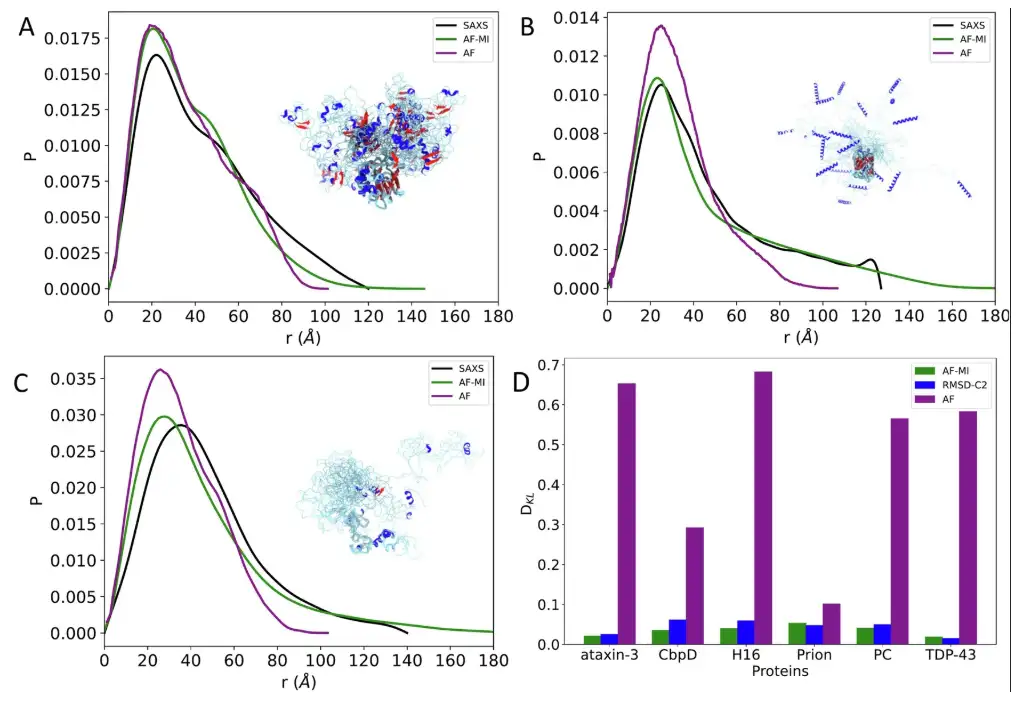
さらに研究者らは、CbpD、H16、PCという3つのタンパク質も研究し、その結果、いずれの場合も、実験値と逆計算値による残基間距離分布の一致は非常に良好です。また、下の図 D に示すように、これは AlphaFold タンパク質構造データベースから直接取得された AlphaFold 単一構造に比べて大幅に改善されています。
最後に、CALVADOS-2 法と比較すると、AlphaFold-Metainference は 6 つのタンパク質のうち 4 つ (アタキシン-3、CbpD、H16、PC) で優れたパフォーマンスを発揮し、残りの 2 つ (TDP-43 およびヒトプリオンタンパク質) では同等の構造アンサンブルを生成しました。下の図の通りです。
ディープラーニングに基づく無秩序タンパク質の予測の進歩
過去数年間、AlphaFold は主に折り畳まれたタンパク質の静的構造を予測するために使用されてきましたが、科学研究コミュニティからの批判も受けてきました。この研究は、AlphaFold が無秩序なタンパク質構造の予測にも潜在的な応用上の利点があることを間違いなく確認し、無秩序なタンパク質構造の予測に新たな研究方向も提供しています。
実際、AIと生命科学の密接な統合により、無秩序なタンパク質構造の予測については多くの議論が行われてきました。AIを活用して生命の謎を解明することも、現代の生命科学の分野では主流の方法となっている。
たとえば、以前 Current Opinion in Structural Biology に掲載された記事では、本質的に無秩序なタンパク質 (IDP) の研究におけるディープラーニングの応用の進歩について議論し、無秩序なタンパク質の予測と立体配座の集合体の特徴付けを促進する上でのディープラーニングの役割について説明しました。
関連研究は、「本質的に無秩序なタンパク質のためのディープラーニング:予測の改善から立体構造集団の解読まで」というタイトルで発表されました。
* 紙のアドレス:
https://www.sciencedirect.com/science/article/pii/S0959440X24001775
偶然にも、デンマークのコペンハーゲン大学の研究チームが、Nature 誌に「ヒトの本質的に無秩序なプロテオームのコンフォメーション アンサンブル」と題した無秩序タンパク質研究に関する記事を発表しました。この記事では、IDP の無秩序領域、コンフォメーション アンサンブル、および関連特性を予測するためのさまざまなディープラーニング手法の使用について説明されており、これには前述の AlphaFold などのディープラーニング手法のほか、タンパク質言語モデル、生成的敵対ネットワークなどが含まれます。
*論文の宛先:
https://www.nature.com/articles/s41586-023-07004-5
AIの急速な発展により、人生の真の意味に対する理解が加速していることは間違いありません。かつてイギリスの科学者ジョン・ケンドリューは、X 線結晶構造解析法を使って最初のタンパク質構造を解明するのに 12 年を要しました。現在、AlphaFold は数億のタンパク質の折り畳みの謎を解明するのにわずか数年しかかかりません。将来、無秩序なタンパク質構造の予測をマスターできないと断言できる人がいるでしょうか。








