Command Palette
Search for a command to run...
Natureサブマガジンに掲載されました!北京大学チームがAIを利用して新型コロナウイルス/エイズ/インフルエンザウイルスの進化方向を予測し、精度を向上 67%
2019 年 12 月、SARS-CoV-2 ウイルスによって引き起こされるこの病気は、わずか 1 か月で 1,000 人を超え、急速に世界中に広がりました。
ウイルスのさらなる蔓延に対抗するため、我が国は2021年初頭にすべての人を対象とした無料ワクチン接種政策を開始しました。しかし、ワクチンの恩恵にもかかわらず、この健康危機はますます複雑になっています。これは、SARS-CoV-2 ウイルスが変異を続けているためです。ワクチンによって生成される免疫圧力や環境条件の変化に徐々に適応し、武漢で最初に発見された「ウイルス株」はとうの昔に消滅し、さまざまな変異株に取って代わられ、新たな感染の波を引き起こし続けており、その影響は今後も続くだろう2023年以降まで。
偶然にも、つい最近、インフルエンザウイルスの陽性率が上昇し続けており、多くの人が知らず知らずのうちにインフルエンザA型(以下、インフルエンザA型)に感染しています。SARS-CoV-2ウイルスと同様、インフルエンザA型も感染力が非常に強いです。変異速度が速いため、同じ季節に複数の亜型のウイルスが出現する可能性があり、短期間に感染が繰り返されるリスクも高まります。
ウイルスの進化方向の予測は、予防と制御、ワクチンと薬剤の設計にとって重要であることがわかります。しかし、突然変異はウイルス進化の基礎であり、その高度なランダム性により、通常は非常に少数の突然変異だけがウイルスの適応性を「ほんの」高めることができ、この陽性サンプル(有益な突然変異)と陰性サンプルの不均衡の問題が生じます。サンプル (有害な変異) このため、ウイルスの稀な有益な変異を予測できる深層学習モデルをトレーニングすることが非常に困難になります。同時に、ウイルスは少数の部位のみで変異を起こすことが多いため、変異による弱い分子内相互作用の変化をニューラルネットワークが直接捉えることが難しく、モデリングにも問題が生じます。
この点に関して、北京大学情報工学院のTian Yonghong教授とChen Jie准教授は、広州国立研究所のZhou Peng研究員とともに、博士課程の学生Nie Zhiweiと修士課程のLiu Xudongに、問題を再検討するよう指導した。ウイルスの進化を予測し、進化駆動型ウイルス変異を予測するフレームワーク E2VD を提案しました。このフレームワークは、SARS-CoV-2 ウイルス、インフルエンザ (インフルエンザ ウイルス)、ジカ (ジカ ウイルス)、および HIV (エイズ ウイルス) の進化の方向性を予測することができ、新しいウイルス感染症に対する人間の反応速度を大幅に向上させ、ワクチンと迅速なワクチンを提供します。薬剤の最適化。
この研究は、「ウイルス変異ドライバー予測のための統合された進化駆動型深層学習フレームワーク」というタイトルで、2025 年 1 月 17 日に「Nature Machine Intelligence」に掲載されました。

用紙のアドレス:
https://www.nature.com/articles/s42256-024-00966-9
論文アドレス: 公式アカウントをフォローし、バックグラウンドで「Virus Evolution」に返信すると完全な PDF が入手できます
オープンソース プロジェクト「awesome-ai4s」は、100 を超える AI4S 論文の解釈をまとめ、大規模なデータ セットとツールを提供します。
https://github.com/hyperai/awesome-ai4s
データセット: UniRef90 事前トレーニング データ セットおよびウイルスの深い突然変異スキャン データ セット
ウイルスは進化の過程で新しい突然変異を生成し続け、それらを選択的に蓄積します。そのため、進化シナリオのタンパク質言語モデルには強力なゼロサンプル一般化機能が必要です。つまり、目に見えない突然変異の状況に対処できなければなりません。これを達成するには、研究チームは、タンパク質言語モデルの事前トレーニング データセットとして UniRef90 を選択しました。 UniRef90 には、モデル トレーニングの初期段階でのパフォーマンスに悪影響を与えることなく、豊富なシーケンス レベルの進化情報が含まれています。この豊富な進化情報により、事前トレーニング プロセス中にモデルを十分なタンパク質ファミリー シーケンス サンプルにさらすことができるため、ゼロサンプル汎化能力が向上します。
さらに、ウイルスの突然変異によって引き起こされる進化的適応度ランドスケープのモデル学習をサポートするために、研究チームは、さまざまなウイルスのオープンソースのディープミューテーションスキャンデータセットを使用しました。
モデル アーキテクチャ: 進化にインスピレーションを得たユニバーサル アーキテクチャ デザイン
研究チームは、「弱い変異増幅」と「希少有益変異マイニング」の設計に基づいて、進化駆動型ウイルス変異推進力予測フレームワークE2VDを提案した。下の図aに示すように、主に3つのモジュールが含まれており、それらは、タンパク質配列のエンコーディング、ローカルとグローバルの依存結合、およびマルチタスクの焦点学習です。
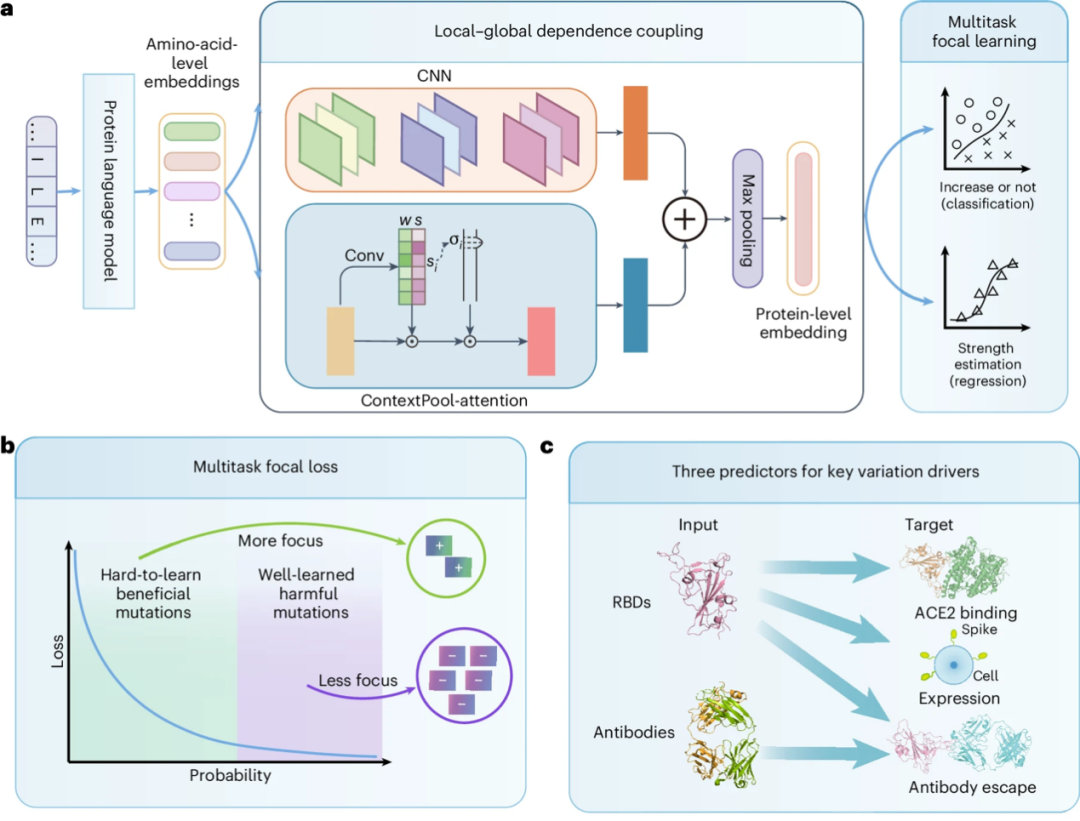
* 初め、タンパク質配列コーディングモジュールでは、研究チームはウイルス進化のためにカスタマイズされたタンパク質言語モデル(タンパク質言語モデル)を独自にトレーニングしました。これにより、ウイルスタンパク質配列の特徴を正確に抽出できます。
※第二に、ローカルとグローバルの相互作用依存性融合モジュールでは、研究者らは畳み込みニューラル ネットワーク (CNN) を使用して、変異と隣接するアミノ酸間の相互作用依存性を捕捉し、学習可能な動的注意メカニズムを設計して、変異が存在する場所のモチーフレベルの表現を構築しました。長距離の相互作用はネットワークに依存します。この設計は、バリアント内の全体的な突然変異が少ないことによって引き起こされる弱い影響を捉えるのが難しいという問題を効果的に解決します。
* それから、マルチタスク集中学習モジュールでは、マルチタスク学習の利点と困難なサンプルマイニング戦略が組み合わされ、マルチタスクトレーニングのパラメーター共有を通じてウイルス変異適合性に対するモデルの予測パフォーマンスが向上します。
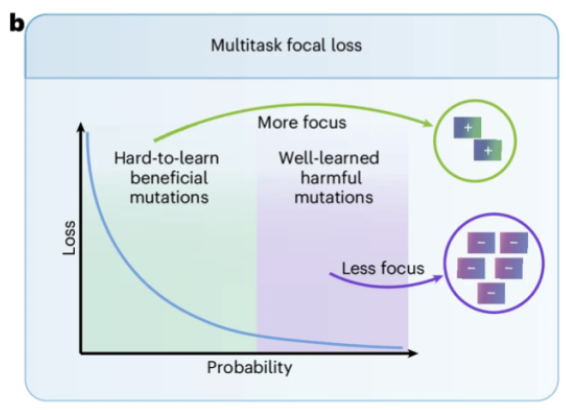
さらに重要なのは、上の図 b に示すように、チームは新しいマルチタスク焦点損失関数 (マルチタスク焦点損失) を設計しました。これにより、モデルは、トレーニング中に効果的に学習することが難しいまれな有益な変異にさらに注意を払うようになります。したがって、まれな有益な突然変異 (つまり、ハード サンプル) の予測パフォーマンスが大幅に向上します。
さらに、上の図 c に示すように、E2VD 予測フレームワークは、さまざまなウイルス適合性予測タスクの入力と出力を柔軟に調整できます。たとえば、変異による結合親和性の変化を予測するには、ウイルス配列のみを入力して、変異による抗体回避能力の変化を予測 ウイルス配列と抗体配列を同時に入力できるため、ウイルスの種類や株を超えた高精度な進化予測を統一的なアーキテクチャ上で実現します。
具体的には、この研究では、SARS-CoV-2 ウイルス、インフルエンザ (インフルエンザ ウイルス)、ジカ (ジカ ウイルス)、HIV (エイズ ウイルス) に関連する予測タスクに E2VD フレームワークが使用されました。
* SARS-CoV-2 のタスクには、結合親和性、発現、抗体エスケープの予測が含まれます。これら 3 つのタスクは、ウイルス変異の主要な要因です。
* インフルエンザ、ジカ、および HIV ウイルスのタスクは、モデルの汎化能力を分析するために、突然変異によって引き起こされる適応度の影響を予測することです。
実験結果:E2VDは67%による有益な変異の予測精度を向上させ、優れた汎化性能を有する
E2VD はウイルスの進化パターンを正確に捕捉し、有益な変異の予測精度を向上させることができます 67%
チームは、進化シナリオ用にカスタマイズされたタンパク質言語と主流のタンパク質言語モデルの予測パフォーマンスを比較しました。その結果、チームのカスタマイズされたタンパク質言語モデルが、最小数の 3 億 4,000 万のモデル パラメーターで最高の予測パフォーマンスを達成し、さらには 3 億 4,000 万個のモデル パラメーターを達成したことがわかりました。これは、ESM2-15B よりも 44 倍効果的であり、カスタマイズされた事前トレーニング データセットとトレーニング戦略の有効性を証明しています。
その後、チームはさまざまな主要なウイルス進化の推進力予測タスクにおいて E2VD を主流の手法と比較した結果、E2VD が他の手法を大幅に上回り、7% から 21% までの範囲でパフォーマンスが向上したことが示されました。さらに、さまざまな種類の変異の正確な区別や、まれで有益な変異の正確なマイニングなど、ウイルスの進化パターンを正確に捕捉するE2VDの能力を証明するために、研究者らは複数の実験を実施した。
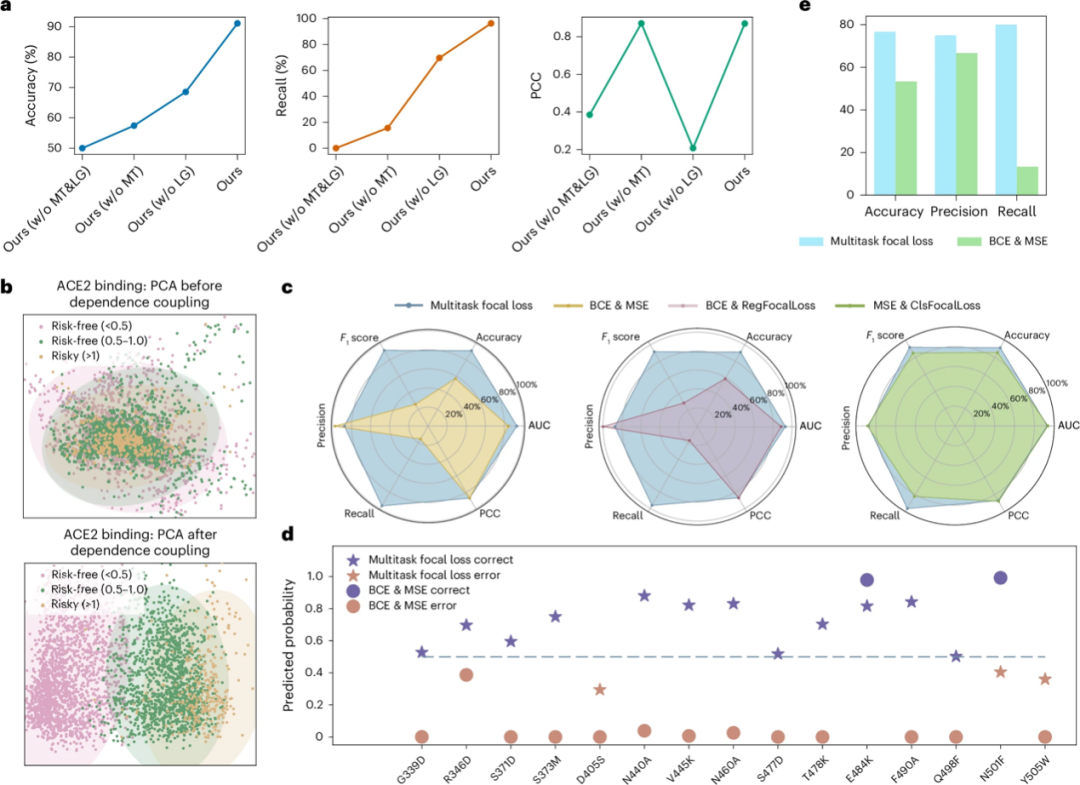
a: MT なしは MT モジュールなしの E2VD を表します。LG なしは LG モジュールなしの E2VD を表します。MT&LG なしは MT&LG モジュールなしの E2VD を表します。
b: 親和性予測タスクで説明されるリスク レベルに結合する 3 つの変異タイプ
d: まれな有益な突然変異を捕捉するためのさまざまな損失の能力
まず、モジュールアブレーション研究を使用して、ローカル-グローバル相互作用依存融合 (LG) モジュールとマルチタスク焦点学習 (MT) モジュールの予測パフォーマンスへの寄与を調査します。上の図aに示すように、この研究では、MTモジュールがウイルス適応において有益な希少な変異をマイニングするのに効果的であることがわかりました(再現率は0から69.63%に増加しました)。 LG モジュールと MT モジュールを組み合わせると、モデルのパフォーマンスがさらに向上し、精度は 91.11%、再現率は 96.3%、相関係数は 0.87 に達します。
チームが提案したマルチタスク焦点損失関数は、予測パフォーマンスを大幅に向上させることができます。まれな有益な突然変異を捕捉する際のマルチタスク焦点損失の能力を評価するために、研究者らは代表的な有益な突然変異と有害な突然変異を選択してテストセットを形成した。
* 有益な変異の予測については、上の図 d に示すように、E2VD はまれな有益な変異の予測精度を 13% から 80% に向上させ、飛躍的な精度の向上を実現します。これにより、ウイルスの進化に重要なまれな有益な変異を正確かつ効率的にマイニングできます。 。
* 有害な変異の場合、マルチタスク フォーカス損失は従来の BCE&MSE と同様に機能します。これは、BCE&MSE ではモデルが希少な有益な変異を学習することができず、モデルがすべての変異を有害な変異として予測する傾向があるためです。
以下の図 b に示すように、研究者らは主成分分析 (PCA) を使用して、インフルエンザ、ジカ、HIV の 3 種類の変異について次元削減の視覚化を実行しました。その結果、LG モジュール処理後、さまざまな変異の特性が明確に区別されることが示されました。 . 明確な境界線を分離します。これは、LGが分子内相互作用ネットワークを捕捉して再構築することにより、さまざまな種類の突然変異に対するE2VDの感受性を高め、それによってウイルスの進化的適応性をよりよく理解できることを示している。
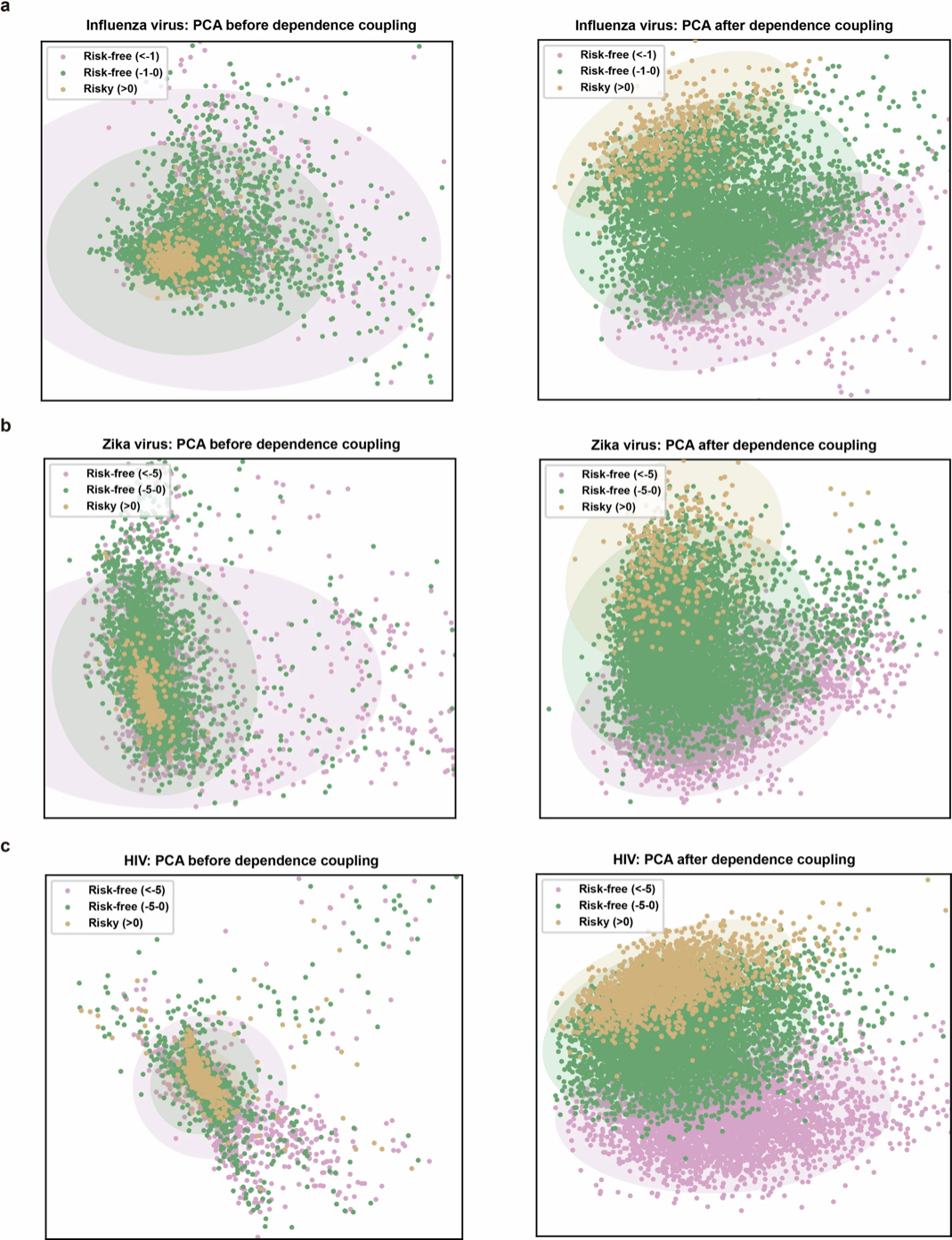
E2VD は優れた一般化パフォーマンスを備えており、ウイルスの種類や株全体を予測できます。
ウイルスは選択圧のもとで進化を続け、例えば最近注目を集めているインフルエンザウイルスには複数の型があり、季節変動を示します。したがって、複雑なウイルスの進化傾向に対処するには、モデルの一般化能力が重要です。研究者らは、同じウイルス、異なる株、異なる種類のウイルスを予測するモデルの能力を評価する「順序ペア比率 (OPP)」を提案しました。能力。
* OPP は、すべての変異ペアのうち正しく予測された変異ペアの割合を表します。OPP 値が大きいほど、予測された適応度ランドスケープがより無秩序であり、ウイルス変異ドライバーの相対的な順序を予測するモデルの能力がより理想的であることを示します。
以下の図 b に示すように、株間結合親和性予測タスクについて、研究者らは 6 つの異なる株の OPP とすべての株の混合データ (All) を評価しました。その結果、さまざまなケースで E2VD が他の株を大幅に上回っていることがわかりました。方法。以下の図 c に示すように、E2VD は、発現レベルの予測タスクにおいて、大部分の菌株において他の方法よりも優れたパフォーマンスを発揮します。全体として、E2VD は分布外株に対して最先端の手法を包括的に上回っており、高い汎化パフォーマンスを示しています。
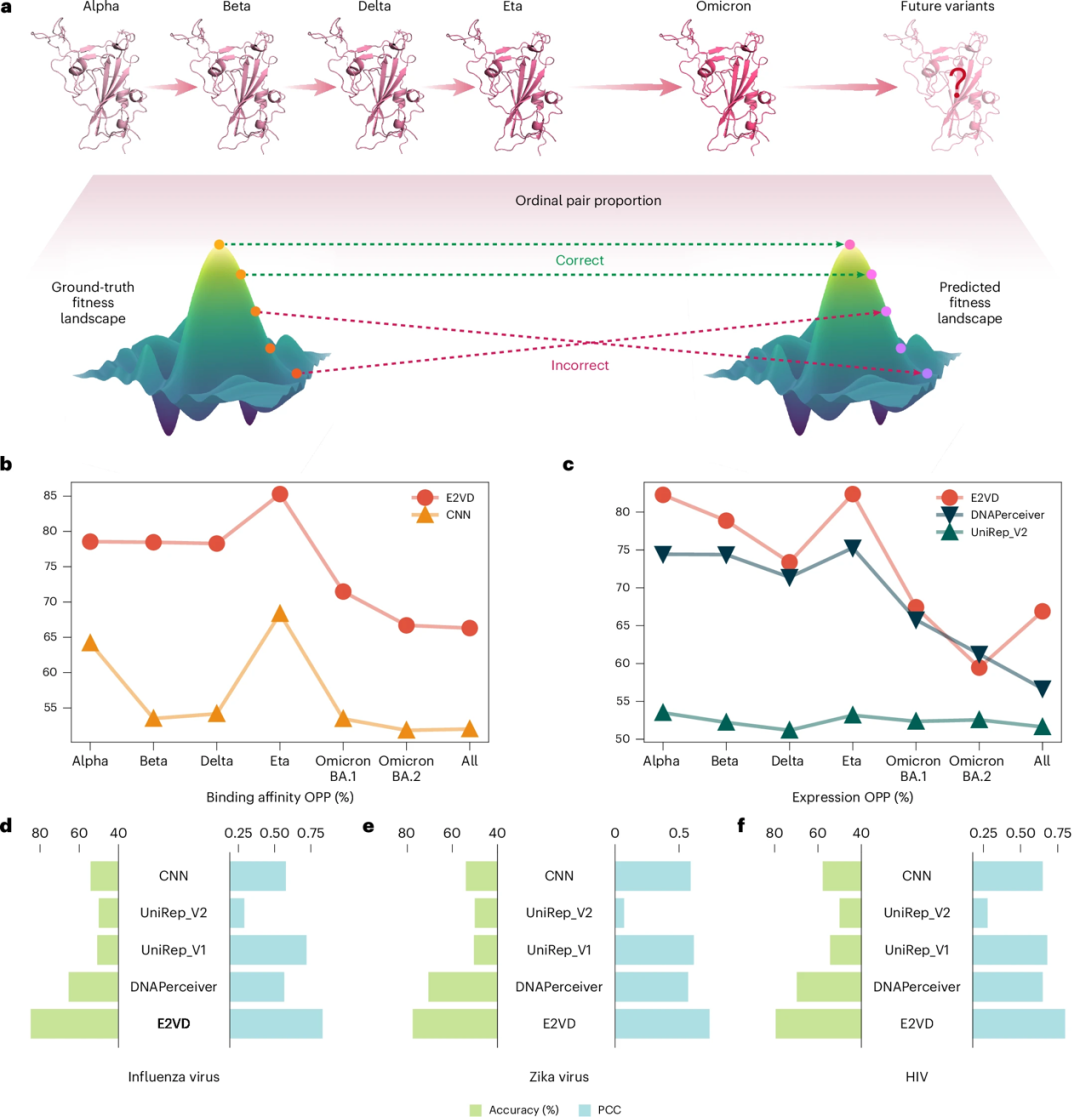
* b、c: E2VD はさまざまなウイルス株の OPP を予測します; d、e、f: E2VD はさまざまな種類のウイルスのパフォーマンスを予測します
上の図 d、e、f に示すように、クロスウイルス型予測の観点から、研究者らは、E2VD が新型コロナウイルス、ジカウイルス、インフルエンザウイルス、HIV に対して理想的な汎化能力を示し、他の手法を上回っていることを発見しました。将来的には、より感染力の高いウイルスが含まれるようにさらに拡張される可能性があります。
AIはウイルスの進化を予測する大きな可能性を秘めています
上記の研究は、進化理論の観点からウイルス進化予測の問題を再検討し、さまざまなウイルスの種類と株に適した一般的な進化予測フレームワーク E2VD を構築しました。このフレームワークは、複数のウイルス変異ドライバー予測において優れた予測性能と精度を示しました。一般化能力により、ウイルスの進化傾向を予測することが可能になります。さらに、E2VD を柔軟にカスタマイズして組み合わせることで、さまざまなスケールでの進化傾向の予測も実現できます。
* まず、E2VD はパンデミックにおけるウイルス進化の経路を説明でき、ウイルス株が蔓延する理由とその背後にある分子メカニズムを理解するのに役立ちます。
* 第 2 に、E2VD は仮想の深度突然変異スキャン シミュレーションと組み合わせることで、80% のヒット率を達成する可能性のある高リスクの突然変異を予測できます。
* 最後に、E2VD はパンデミック規模の大進化軌道予測も実現し、現実世界でのウイルスの進化経路を再現することで、ウイルスの進化メカニズムの解釈に理論的な裏付けを提供します。
将来的にチームは、E2VDをワクチンおよびタンパク質医薬品設計プロセスと組み合わせて、設計の効率と制御性を向上させることを計画しており、これはウイルス予防と医薬品設計にとって非常に重要です。
この研究の著者は、北京大学情報工学部のTian Yonghong教授とChen Jie准教授、そして彼らの指導を受けた博士課程の学生Nie Zhiweiと修士課程の学生Liu Xudongであることは言及する価値がある。チームは引き続き次のことに焦点を当てている。ライフサイエンスのための AI 分野の研究「ウイルスの進化に先駆けて - 人工知能シミュレーションによる将来の高リスク新型コロナウイルス変異種の予測」が 2022 年の最終候補者に選ばれました。毎年恒例の「ゴードン・ベル新型コロナウイルス感染症特別賞」(ゴードン・ベル賞は、ハイパフォーマンス・コンピューティング・アプリケーションの分野における世界最高の学術賞)。
チームはウイルス進化予測の分野で深い蓄積を持っています。 2023年7月、チームは「進化の先を走る—将来の高リスクSARS-CoV-2変異種を予測するためのAIベースのシミュレーション」をThe International Journal of High Performance Computing Applicationsに発表した。具体的には、研究者らは大規模なタンパク質言語モデルを事前トレーニングし、結合親和性と抗体エスケープ予測に基づいたハイスループットのスクリーニング方法を構築しました。これは、SARS-CoV-2 RBD 変異シミュレーションに関する初の研究であり、このモデルは懸念される 5 つの変異株の RBD 領域の変異を特定し、数百万の潜在的な変異株を数秒以内に排除することに成功し、流行の予防と制御のための情報を提供しました。 「AI+HPC」(人工知能 + ハイパフォーマンス コンピューティング)パラダイムの技術的手段。
論文リンク:
https://journals.sagepub.com/doi/abs/10.1177/10943420231188077
さらに、チームは生命科学のための一連の基本モデルを開発しました。酵素工学にとって重要な「酵素-基質」相互作用予測タスクを例として、チームは2024年12月にプレプリント論文を発表し、酵素-基質相互作用予測のための多目的の進歩的条件付き深層学習を提案しました。
論文リンク:
https://www.researchsquare.com/article/rs-5516445/v1
具体的には、酵素と基質の相互作用のモデリングを 2 段階の学習プロセスに分離することにより、2 つの条件付きネットワークがそれぞれ酵素反応の特異性と重要な触媒相互作用情報を導入するように設計され、それによってタンパク質から抽出される特徴潜在空間が促進され、一般的な小分子の分野は徐々に触媒センシングの分野に移行しました。このモデルは、さまざまな下流タスクにわたって一貫して最先端の手法を上回ります。さらに、提案された条件付きネットワークは、無視できる追加の計算オーバーヘッドで酵素触媒作用の基本パターンを暗黙的に捕捉します。この条件認識メカニズムによってサポートされるこのモデルは、構造情報を必要とせずに、活性部位を正確に特定し、重要な触媒相互作用に関与する酵素残基と基質官能基を効率的かつ低コストの方法でマイニングすることができます。
人工知能の支援を受けて、チームはライフサイエンス向けAIの関連分野での徹底した研究をさらに推進し、ウイルス予測、タンパク質創薬、ワクチン開発などの可能性をさらに広げていきます。さらなる成果が期待されます。
参考文献:
https://www.who.int/
https://news.pku.edu.cn/jxky/90d276ae5f8441849fd04372fd872154.htm
https://news.pkusz.edu.cn/info/1003/8711.htm









