Command Palette
Search for a command to run...
南開大学のZheng Wei教授:AlphaFoldは完璧ではなく、学術界にはまだ「コーナーでの追い越し」の可能性がある

近年、ディープラーニングなどのAI技術の支援により、タンパク質の構造予測の分野は急速に発展し、2024年10月にはDeepMind社のデミス・ハサビス氏とジョン・M・ジャンパー氏がAlphaFoldで2024年のノーベル化学賞を受賞した。これは、AlphaFold がかけがえのないものであり、他の優れたアルゴリズムがまだ探索する価値があることを意味します。
生放送シリーズ「Meet AI4S」第6回では、HyperAI は幸運にも、南開大学統計データサイエンス学部の教授である Zheng Wei 教授を招待することができました。「AlphaFold3の王座はまだ安定せず、アカデミアからの逆転~深層学習による生体高分子の立体構造予測とその相互作用~」をテーマに、AlphaFoldの限界と今後の最適化の方向性を皆さんに共有しました。学術コミュニティと研究する価値のあるアルゴリズムと研究トピックは何ですか?
* 公開アカウントをフォローし、「Meet AI4S Issue 6」に返信するとスピーチ PPT を入手できます
HyperAI Super Neural は、当初の意図に反することなく詳細な共有を編集および要約しました。以下は講演の書き起こしです。
AlphaFold の制限事項
タンパク質は生命活動の根幹であり、タンパク質の立体構造を予測することは生体機能を理解する上で極めて重要です。 DeepMind が発売した AlphaFold 2 はタンパク質構造予測を新たなレベルに引き上げましたが、これは AlphaFold 2 のエンドツーエンド フレームワークがタンパク質構造予測の問題をすべて解決したことを意味するものではありません。
まず、AlphaFold 2 自体を例に挙げると、依然として多くの制限があります。
*精度を改善する必要があります
公式レポートによると、AlphaFold 2 は 90% 以上の精度で構造を予測できますが、実際のタスクではこれほどの精度に達することはできません。
* マルチドメインのタンパク質構造予測には限界があります
AlphaFold 2 はシングルドメインタンパク質の予測には優れていますが、複雑なマルチドメインタンパク質の場合、ドメインは比較的柔軟であり、予測精度は良くありません。
* タンパク質の複合体構造の予測には限界があります
タンパク質が機能するには、他のタンパク質と複合体を形成する必要があることがよくありますが、AlphaFold 2 の初期バージョンではこの問題に対処していませんでした。
※RNA構造予測、RNA-RNA、タンパク質-RNA構造予測は限定されます
上で述べたように、これらの問題は初期バージョンでは対処されていませんでした。
*タンパク質の動態/構造変化の予測は限定的
実験的な解析手法では通常、ある瞬間の構造状態しか捉えることができませんが、タンパク質は生体内に静的に存在するわけではなく、異なる時点での構造は異なる可能性があります。これらの問題は、AlphaFold 2 ではまだ解決されていません。
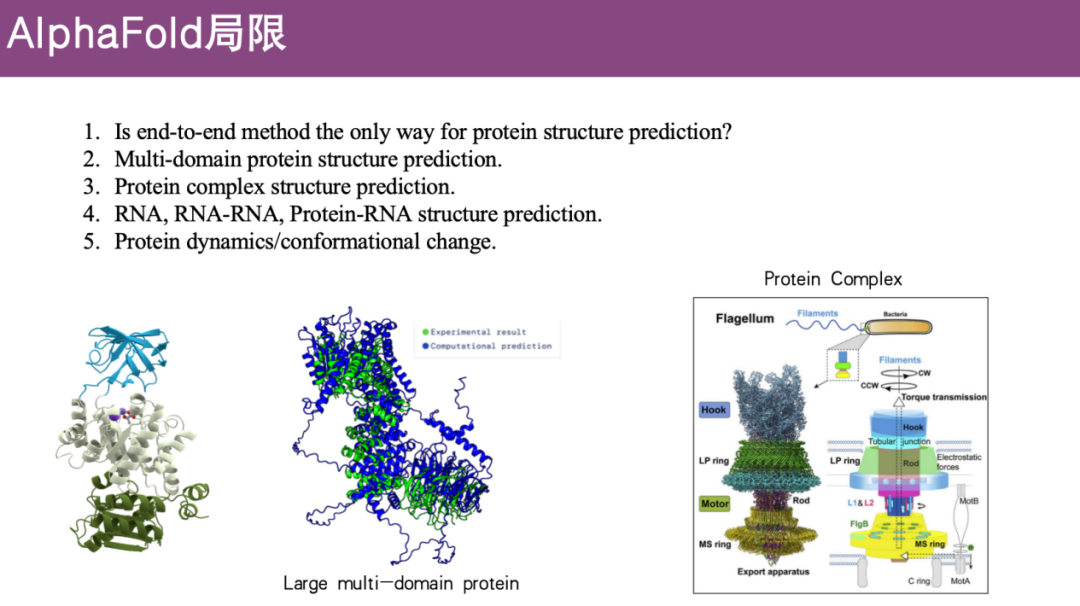
さらに、DeepMind は AlphaFold 3 を反復しており、タンパク質モノマーの構造予測において優れたパフォーマンスを発揮することは誰もが知っていますが、複合体、核酸、および小分子の予測精度はまだ改善の必要があります。したがって、次世代の AlphaFold では、他の機能を備えた予測モジュールが追加される可能性があります。たとえば、既存のモデルが主に静的構造を扱うために使用されていることを考慮すると、タンパク質のアロステリーを予測するために分子動的プロセスが探索されます。あるいは、タンパク質設計の分野が関与し、予測プロセス全体が逆転する可能性もあります。
したがって、AlphaFold があっても、学術界全体としてはまだやるべきことがたくさんあります。
AlphaFold 以外に、検討する価値のある方法はありますか?
これまで、タンパク質の三次元構造を解析する主な方法は、X 線、核磁気共鳴 (NMR)、および極低温電子顕微鏡でした。タンパク質の構造を実験的に解析することは難しく、コストがかかるため、チームによっては、タンパク質の三次元構造を解析するには数か月から数年かかります。その結果、人々はより経済的でより迅速な方法、つまりアルゴリズムを通じてタンパク質の構造を予測する方法を模索し始めました。
タンパク質は主に英語の文字で表される20種類のアミノ酸で構成されており、アミノ酸分子にも多くの原子が含まれていることがわかっています。したがって、タンパク質の構造予測問題は次のように要約できます。これらの文字で構成されるアミノ酸列を入力し、計算アルゴリズムを使用して、各アミノ酸の各原子の 3 次元空間座標 (x、y、z) を予測します。タンパク質の配列。
タンパク質の構造予測の開発プロセスを通じて、比較モデリングや相同性モデリング、分子動力学シミュレーション (MD)、スレッディング アルゴリズム、デノボ予測、深層学習による構造予測アルゴリズムなど、さまざまな段階でさまざまな代表的なアルゴリズムが登場してきました。連絡先グラフなど主な紹介文は以下の通りです。
* 比較モデリングまたは相同性モデリング
この方法は生物進化の原理に基づいており、配列類似性が高い場合、タンパク質の構造と機能も比較的類似していると考えられます。したがって、まず未知のタンパク質のアミノ酸配列を取得し、次に配列比較を通じて PDB データベース内で配列類似性の高い解決されたタンパク質構造テンプレートを見つけ、移動またはアライメントを通じて未知のタンパク質の構造を予測することができます。
*PDB データベースには、この分野で解明されているタンパク質の構造が含まれています

※分子動力学シミュレーション
基本的な考え方は、タンパク質のアミノ酸配列に基づいて初期の三次元構造をランダムに生成し、各原子にランダムな座標を割り当て、原子の位置を調整し、その後、その配列に基づいてさまざまな時点でのタンパク質の状態エネルギーを計算することです。事前に構築された物理エネルギー力場。エネルギーが最も低い構造が、合理的なタンパク質の立体構造です。

* スレッディングアルゴリズム
相同性モデリングと同様に、違いは、配列類似性の高いタンパク質構造は類似していることが多いものの、構造が類似しているタンパク質の配列類似性が非常に低い場合があり、そのようなタンパク質の適切なテンプレート情報が PDB データベースで見つからないことです。そこで研究者らは、収集した相同配列に基づいて 2 つのタンパク質プロファイルを整列させ、異なるアミノ酸を整列させる多重配列アライメント (MSA) を使用するプロファイルの概念を提案しました。
つまり、二つのアミノ酸配列が異なっていても、しかし、それらのプロファイルは似ており、それはそれらが似た構造を持っていることを意味します。これを使用してテンプレートを検索します。

* デノボ予測
一部のタンパク質は、データベースにある類似の構造を必ずしも持っているとは限りません。そこで研究者らは、タンパク質配列全体を短いフラグメントに分解し、データベース内でこれらの小さなフラグメントのテンプレートを探し、これらの小さなフラグメントのテンプレートを組み立てて完全な三次元構造にすることで予測を試みようとしました。
具体的には、ワシントン大学の David Baker 教授が Rosetta ソフトウェアを開発しました。その主な原理は、タンパク質配列を多数の小さなフラグメントに分解した後、分子動力学シミュレーションで開発されたエネルギー関数を使用して最適化することです。同様の構造予測は、動的シミュレーションとエネルギー最小化の原則に基づいています。
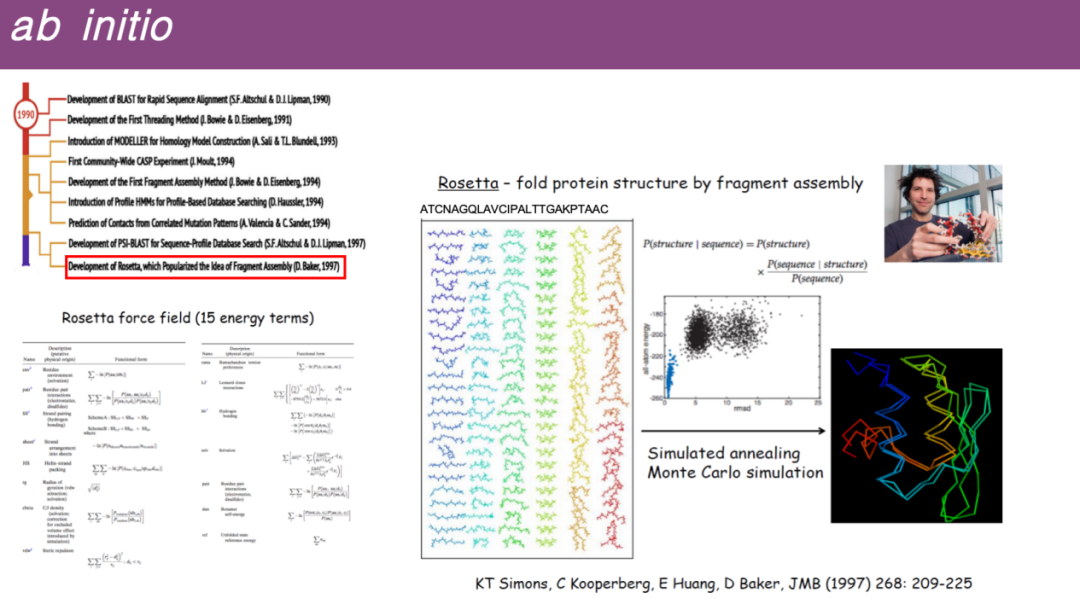
※連絡先図
主なアイデアは、タンパク質の 3 次元構造を 2 次元の図に変換することです。タンパク質の三次元構造情報、つまりすべての空間点の座標位置を使用して、異なるアミノ酸間の距離を計算します。2 つのアミノ酸間の距離が小さい場合、接触が形成されると想定されます。特定のしきい値を超えない場合、この定義を使用して 2 次元構造を圧縮します。さらに、この二次元接触マップからの情報は、タンパク質の三次元構造を再構築することができます。
具体的には、研究者らは深層学習に基づいた多くの手法を開発してきました。その中心となるアイデアは、まず多重配列アラインメント (MSA) を構築し、アミノ酸 i と j のプロファイルの共進化情報を観察することです。酸 それらはしばしば空間内で非常に近くにあり、接触を形成します。続いて、共進化情報を学習用の特徴量として深層学習ネットワークに入力し、タンパク質の接触マップを予測し、タンパク質の三次元構造を復元します。
たとえば、Zheng Wei 教授のチームは以前、CI-TASSER と呼ばれるアルゴリズムを開発しました。これは現在、接触図に基づいてタンパク質構造を予測する一般的な方法です。
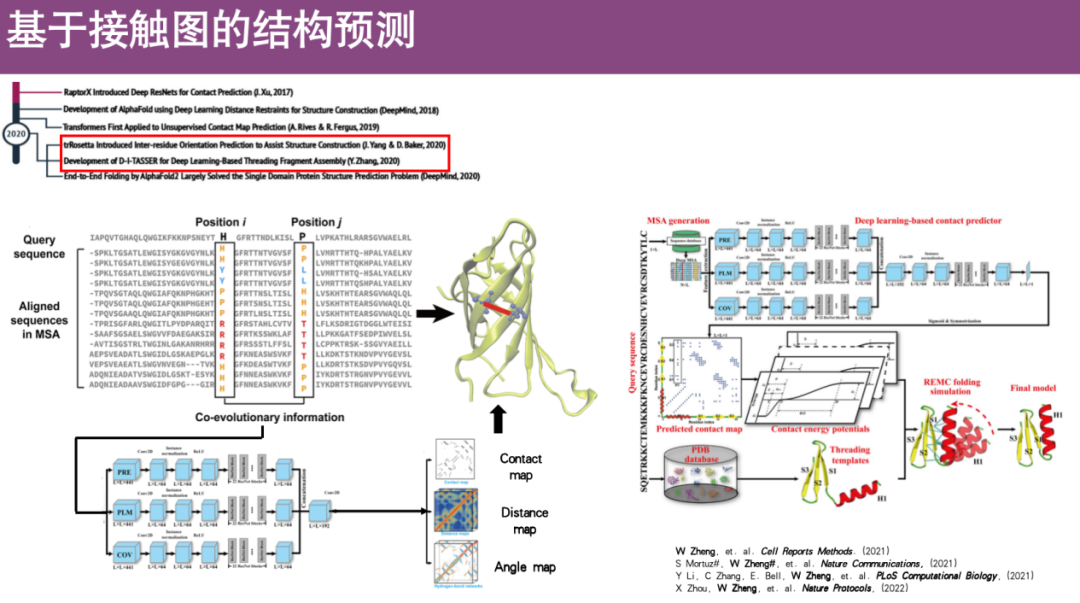
最後に、AlphaFold は、上記のアルゴリズムの多くの基本原理を統合し、タンパク質配列を直接入力し、その後構造を出力できるエンドツーエンドのフレームワークを構築することに成功しました。
チームの成績を例として、学術界で追い抜く機会を探る
タンパク質の構造予測は、生物医学分野に多大な影響を与えます。Zheng Wei教授のチームが現在開発しているアルゴリズムには、未知のウイルスタンパク質(新型コロナウイルス)の構造予測、タンパク質構造解析における低温電子顕微鏡の支援、生物学者によるタンパク質の進化的機能の理解支援、抗体のスクリーニングなどが含まれる。
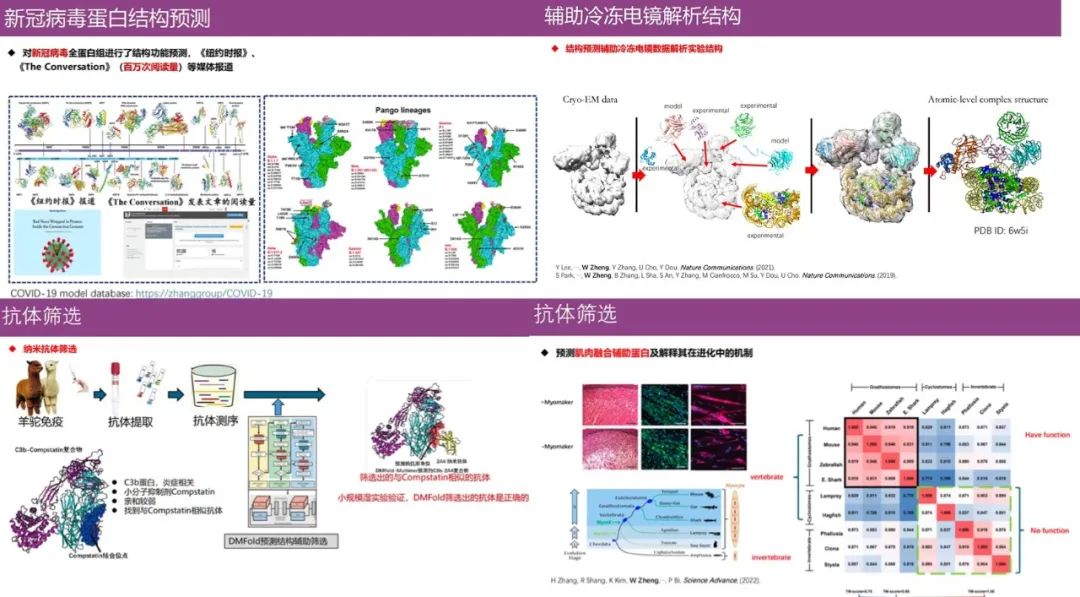
さらに、以下の図に示すように、チームが開発したすべてのタンパク質モノマーおよび複雑な構造の予測アルゴリズムは自動サーバー アルゴリズムに変換され、研究グループの Web サイトで公開されており、そのアルゴリズムは 100 か国以上の 90,000 人以上のユーザーに提供されています。世界中のユーザー、誰でもご利用いただけます。
*プロジェクトの合計アドレス:
https://seq2fun.dcmb.med.umich.edu/DMFold

タンパク質モノマー構造予測法 DI-TASSER
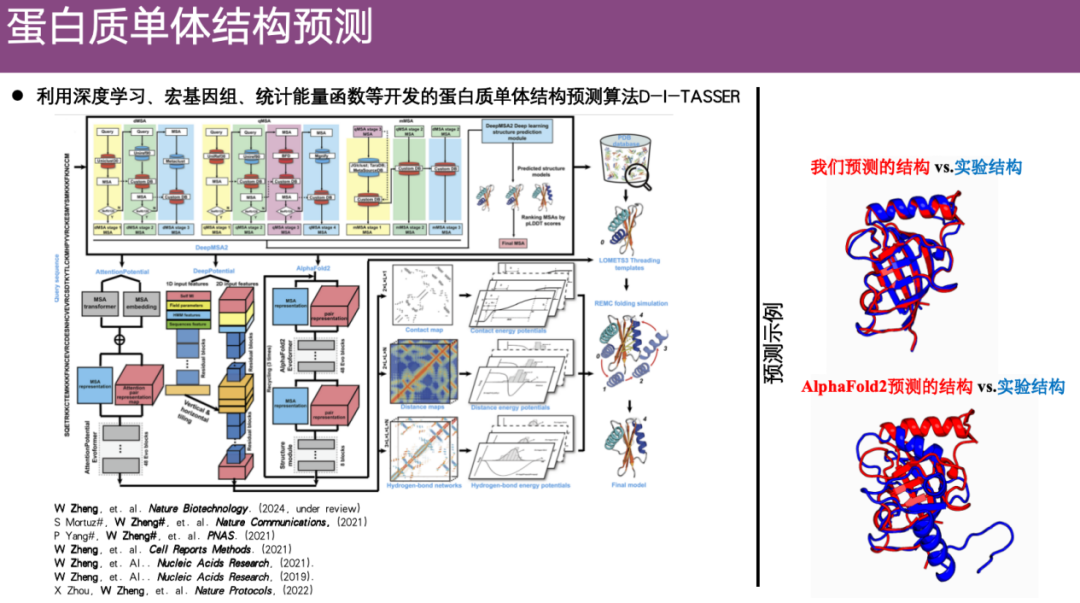
タンパク質モノマーの構造予測の問題は、AlphaFold 2 よりも前から、すでに接触図に基づいた構造予測の研究を行っていました。 AlphaFold 2 の出現後、チームは、AlphaFold 2 によって予測される接触マップなどの空間的制約を、以前に開発されたアルゴリズムに統合できるかどうかを検討し始めました。したがって、空間的制約、メタゲノム、統計的エネルギー関数などに基づいて、チームはタンパク質モノマー構造予測アルゴリズム DI-TASSER を開発し、最適化を行った結果、良好な結果が得られました。
下図の右側のケースに示すように、赤色は DI-TASSER によって予測されたタンパク質構造を示し、青色は実験的に解析された構造を示します。ご覧のように、DI-TASSER によって予測された構造は、実験的に解かれた構造と非常によく似ており、対照的に、AlphaFold 2 で予測された構造と実験構造の間には、アライメント後であっても大きな差異があり、予測精度はわずかに低くなります。
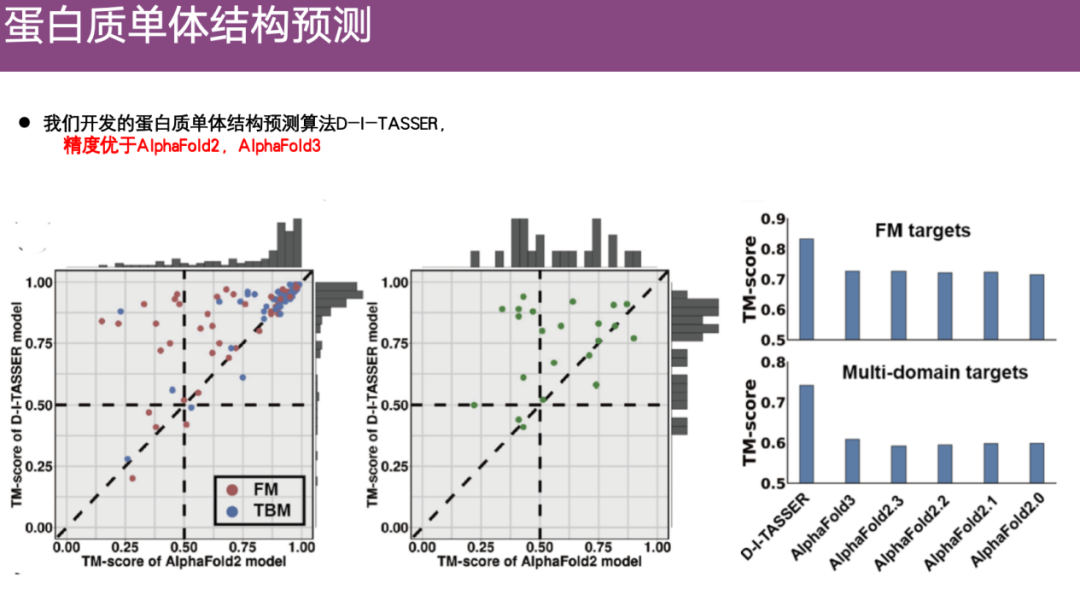
さらに、評価は複数のタンパク質データセットに対して実行されます。下図の右側に示すように、単一ドメインと複数ドメインを予測する場合、DI-TASSER の予測精度は、AlphaFold 2 よりも高く、AlphaFold 3 よりもさらに優れています。
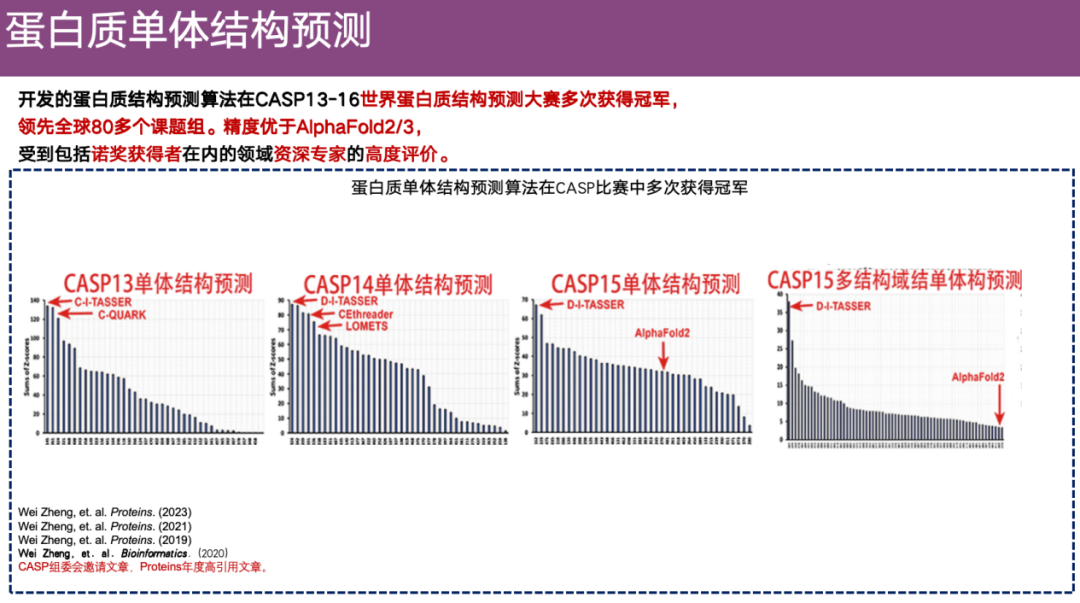
評価の権威を確保するために、チームは内部評価を実施するだけでなく、この分野で権威のあるコンテストである CASP にも参加しました。
CASP コンテストは、この分野ではオリンピックとして知られており、主にタンパク質の構造予測の評価方法を標準化することを目的としています。タンパク質の立体構造予測アルゴリズムには多くの種類があり、各研究室が独自のアルゴリズムを開発しているため、評価データセットや評価方法が異なる場合があり、各研究グループが「自分の手法が世界一正確である」と主張するのが一般的です。この混乱を解決するために、CASP コンテストが誕生しました。
CASP は昨年の時点で 16 回開催され、32 年間にわたって成功裏に開催されており、David Baker 教授のチームや DeepMind チームなど、多くの権威あるチームの参加を集めています。
DI-TASSER とその前身アルゴリズムは、CASP 13 から CASP 15 まで何度も CASP コンペティションに参加しており、CASP 15 ではタンパク質モノマー構造予測の分野で主導的な地位を占めています。DI-TASSER アルゴリズムもマルチドメイン評価に参加し、その全体的な精度は参加したすべての研究グループの精度よりも優れていました。
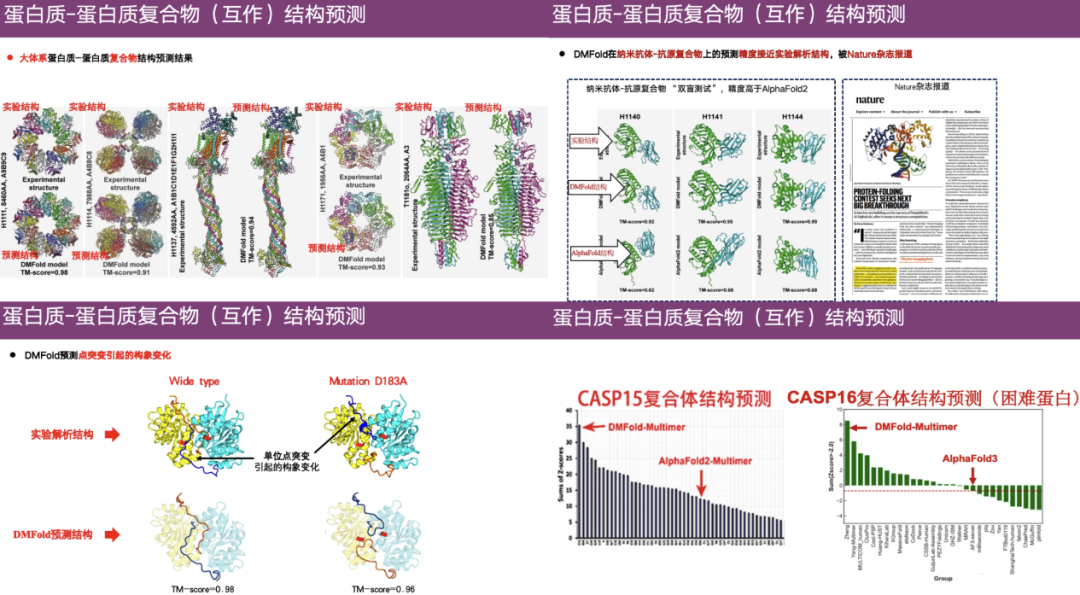
タンパク質複合体構造予測手法 DMFold
複雑な構造予測における主な課題は、共進化情報を通じて分析できる 2 つのタンパク質間の相対的なねじれを予測することです。
たとえば、多重配列アラインメント (MSA) は 2 つのタンパク質のモノマー上に構築され、2 つの MSA はいくつかの接続方法に基づいて 1 つの MSA にマージされ、2 つの MSA 間のアミノ酸の共進化関係を使用して、タンパク質間のアミノ酸の距離も、共進化情報を深層学習フレームワークに統合して、2 つのタンパク質間の相対的なねじれを予測することができます。
この点について、Zheng Wei 教授の研究グループは、より深い多重配列アラインメントを構築するために DeepMSA および MetaSource アルゴリズムを開発しました。さらに、ディープラーニングやメタゲノミクスなどを活用したタンパク質複合体構造予測アルゴリズムDMFoldも開発した。
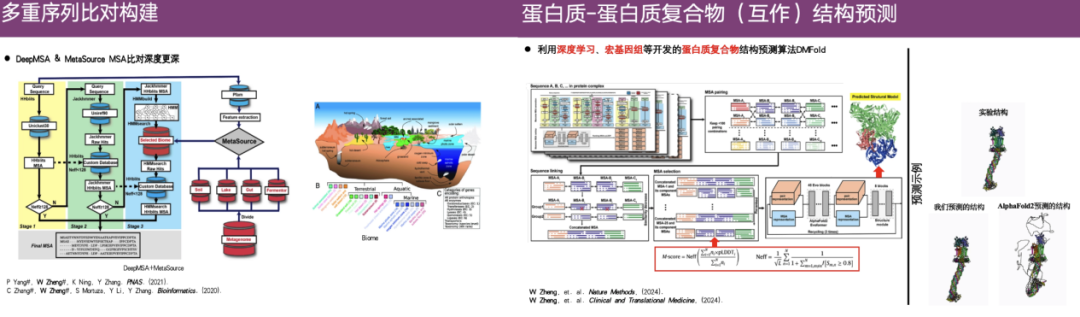
上図の右端のケースに示すように、上が実験解析によって得られた実際の構造、下の左側がDMFoldによって予測された構造、右側がAlphaFold 2によって予測された結果です。 AlphaFold 2 によって予測された構造はカオスであり、異常な触手のような伸長を持っていることがわかります。対照的に、DMFold の予測構造は実験構造と非常によく似ています。これは、DMFold アルゴリズムが化合物構造予測の点で AlphaFold 2 よりも優れていることを示しています。
さらに、DMFold は、大規模システムのタンパク質-タンパク質複合体、ナノボディ-抗原複合体、点突然変異によって引き起こされる構造変化においても高い精度を示します。 CASP15コンペティションでは、 DMFold の全体的なランキングは AlphaFold 2 よりもはるかに高く、CASP 16 では DMFold も AlphaFold 3 よりも優れています。
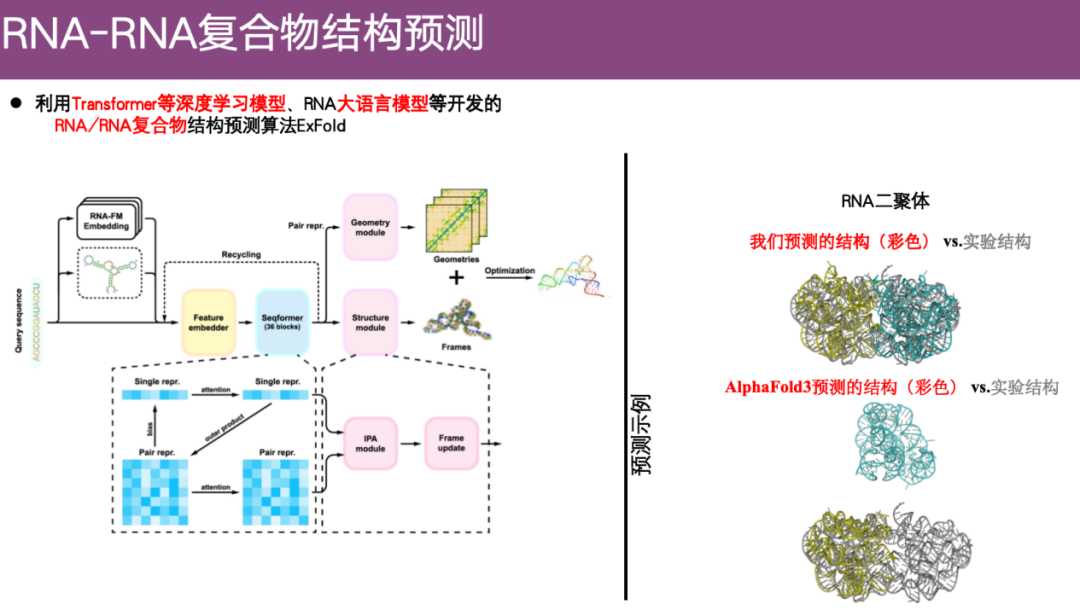
RNA-RNA複合体構造予測法 ExFold
近年ではRNAの構造予測の問題に注目し始めており、例えばTransformerやRNAラージ言語モデルなどの深層学習モデルを用いたRNA/RNA複合体構造予測アルゴリズムExFoldを開発している。
下図の右側のケースに示すように、灰色の部分が実験構造、色付きの部分が予測構造です。ご覧のように、ExFold 法を使用すると、2 つの構造はよく整列しました。対照的に、2 つの RNA 分子間に接触さえ形成されなかったという AlphaFold 3 による予測は、ほぼ完全に間違っていたと考えられます。
また、チームは、下図の左側に示すように、より大きなデータセットを通じて AlphaFold 3 との精度を比較しました。Y 軸は ExFold の予測精度を表します。X 軸は AlphaFold 3 の予測精度を表します。 ExFold の利点は非常に明白であることがわかります。
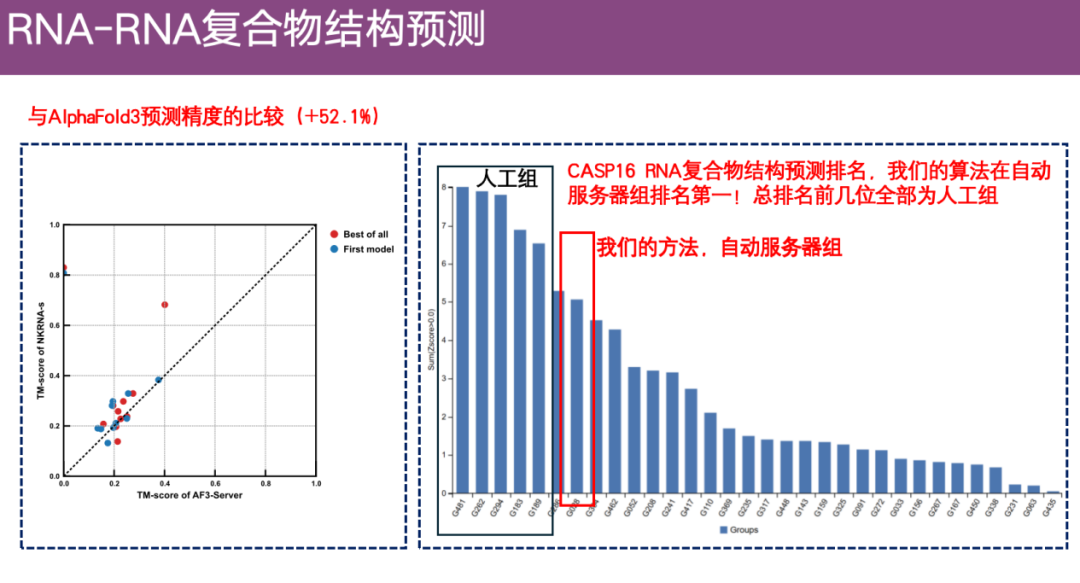
また、CASP16 RNA複合体構造予測コンテストでは、ExFold は全体では 1 位ではありませんが、すべての自動アルゴリズム (サーバー アルゴリズム) の中で最高位にランクされています。
※CASP競技は自動組と手動組に分かれます。自動グループでは、予測結果を 3 日以内に完全に自動で送信する必要があり、手動介入は許可されません。手動グループは 3 週間の猶予期間があり、専門家の経験と手動調整を追加できます。
タンパク質-RNA複合体構造予測手法 DeepProtNA
タンパク質とRNAの複合体構造予測の問題に関して、研究チームは、Transformerなどの深層学習モデルと最近人気のある大規模言語モデルを使用して、新しい構造予測アルゴリズムDeepProtNAを開発しました。
下の右の例に示すように、抗体-RNA複合体において、色はDeepProtNAの予測結果を表し、灰色は実験構造を表します。位置合わせ後、次のことがわかります。DeepProtNA の予測構造は実験構造 (灰色と色の重なり) と非常に一致しており、特に抗体タンパク質と抗原RNAとの接触面における予測精度は非常に高い。対照的に、AlphaFold 3 の予測構造は実験構造とほとんど重なり合わず、予測効果は低いです。
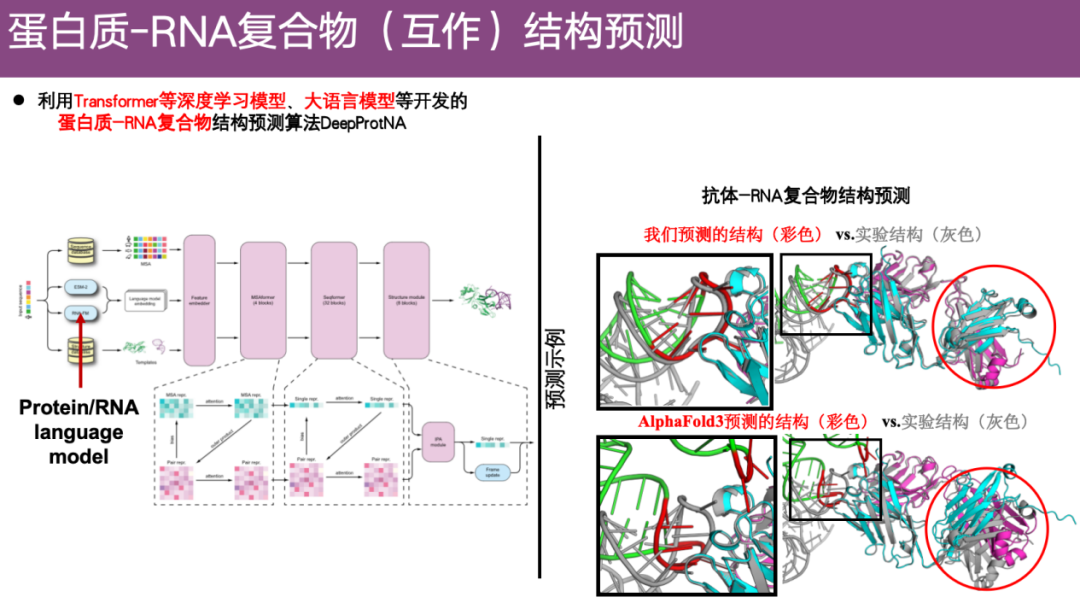
また、DeepProtNA の予測精度は、AlphaFold 3 の予測精度よりも約 7.5 パーセント高く、CASP 16のサーバーグループコンテストで1位にランクイン。
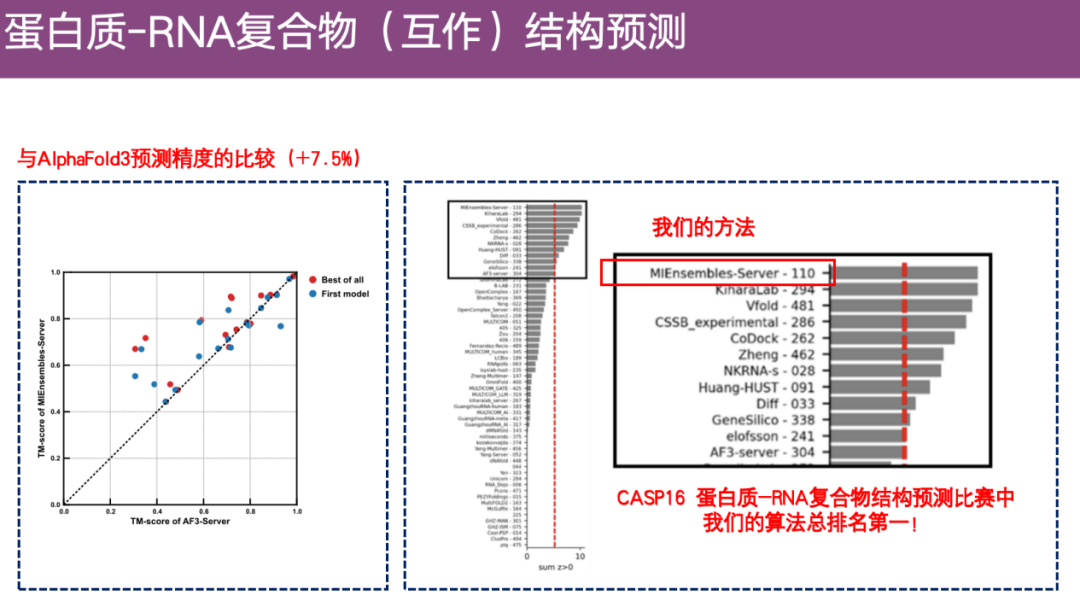
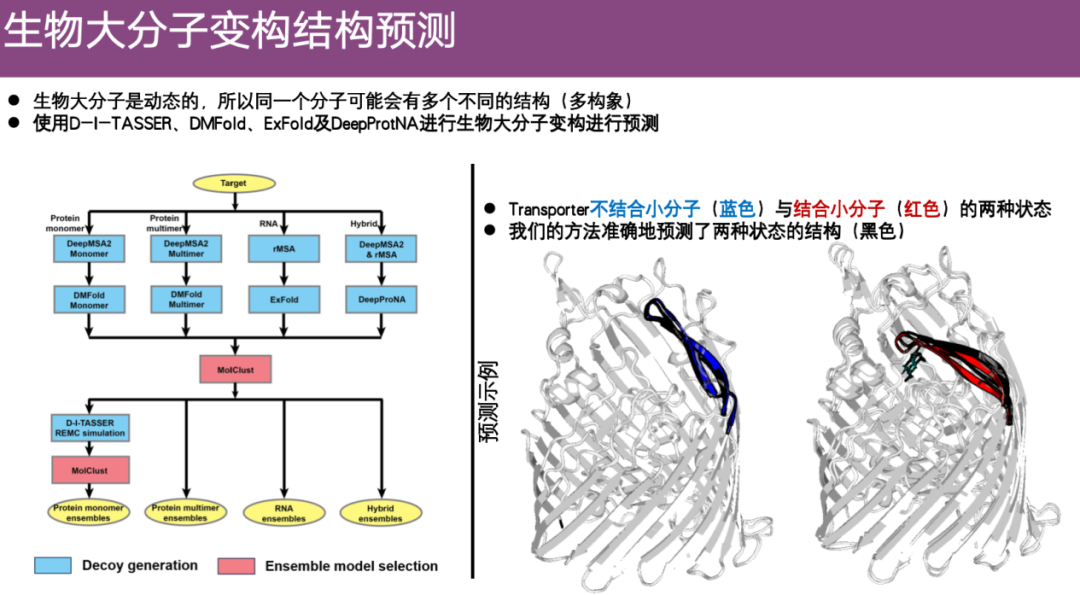
生体高分子のアロステリック構造を予測する手法、EnsembleFold
研究チームは、生体高分子のアロステリック構造予測にも焦点を当てています。高分子多重構造問題の入力はタンパク質配列であり、出力は異なる状態にあるタンパク質の複数のキーフレームです。これは、静的予測アルゴリズムと比較して、複数の異なる構造を単一のアミノ酸配列から予測する必要があることを意味します。これらの構造は、動的プロセス全体を表すキーフレームです。これは、現在の分野で多くの注目を集めているトピックですが、予測するのは困難です。
以前に開発された手法を統合し、高分子のアロステリック問題を最適化することで、研究チームはいくつかのクラスタリング アルゴリズムを開発し、最終的に EnsembleFold と呼ばれるアルゴリズムを形成しました。
下図の右側のケースに示すように、低分子と結合した後のタンパク質の構造変化を示しています。青は低分子が結合していないときの実験構造を示し、赤は結合後のチルトとアロステリーを示します。緑色の小さな分子を結合します。チームは入力されたタンパク質配列に基づいて、2 つの構造 (黒い部分) を予測しました。小分子を組み合わせていない場合の EnsembleFold の予測構造は、実際の構造と非常に一致しています。小分子を組み合わせた後も、EnsembleFold は実験構造によく適合することがわかります。したがって、EnsembleFold は、生体高分子のアロステリック予測において非常に高い精度を示します。
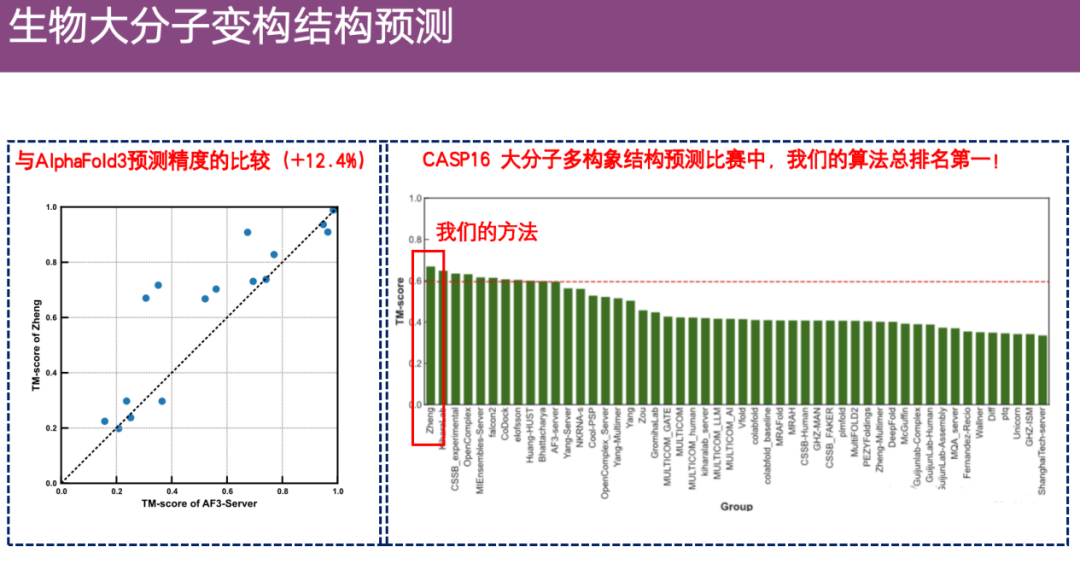
同時に、AlphaFold 3 と比較すると、EnsembleFold の精度が約 12.4% 高いことがわかります。CASP 16 のすべての高分子立体構造コンペティションの中で第 1 位にランクされています。
興味深い例は、研究チームが CASP におけるファージ DNA インテグラーゼのアロステリック プロセスを予測したことです。以下の図に示すように、ファージのアミノ酸配列は P-P' で表され、細菌の遺伝物質配列は B-B' で表されます。ファージ DNA インテグラーゼは、動的プロセスを通じてファージのアミノ酸配列を組み込みます。遺伝物質 P' が細菌の遺伝物質 B に結合して B-P' を形成し、構造が変化します。
研究チームはアルゴリズムを使用して、この多構造変化を予測しました。左側が実験構造を示し、上が未統合状態(立体構造1)、下が統合状態(立体構造2)である。研究チームの予測は、これら 2 つの異なる立体構造を正確に反映していることがわかります。
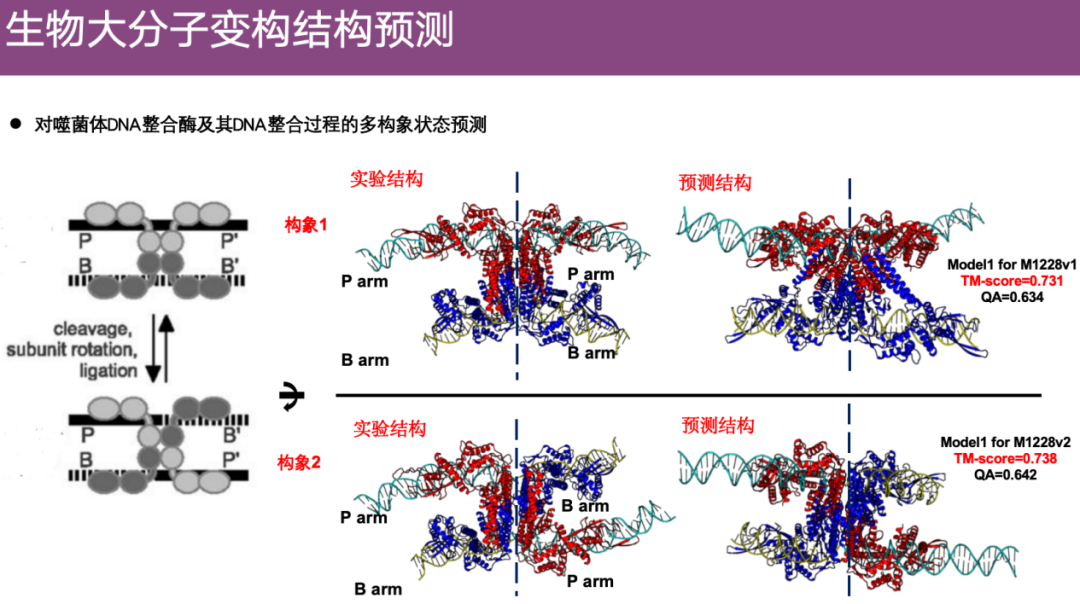
CASP 16 コンテストでは、出場者らは配列情報しか得ておらず、具体的な生物学的プロセスや構造変化の詳細については知らなかったが、鄭偉教授のチームは予測を通じて生物学的プロセス全体を復元することに成功した。試合後の総括では、審判団からも驚きの声が上がった。
研究チームの新メンバーを募集します
南開大学統計データサイエンス学部の鄭偉教授は、タンパク質などの生体高分子の構造、機能、相互作用の予測に長年取り組んでおり、数多くのタンパク質モノマー、タンパク質複合体、タンパク質複合体、タンパク質の開発を主導してきました。 AlphaFold2/3を上回る精度の核酸や複合体、タンパク質核酸複合体構造予測アルゴリズム、構造評価アルゴリズムなどで、世界タンパク質構造予測コンペティション(CASP)の複数のコンテストで優勝を重ねています(CASP13-) 16) 、世界中の 80 以上の学術/産業界の研究グループを主導しています。
彼が勤務する南開大学統計データサイエンス学部のバイオインフォマティクスチームは新メンバーを募集している。計算構造生物学、生物情報学、データサイエンスに興味がある方は、修士号、博士号、ポスドクのいずれであっても、Zheng Wei 教授のチームに参加することを大歓迎です。
興味のある学生は、次の方法で Zheng Wei 教授に連絡してください。
※メール:[email protected]
* WeChat: 18622152765









