Command Palette
Search for a command to run...
NeurIPS 2024に選出されました! Westlake University は、AlphaFold 3 をさらに補完するユニバーサル分子逆フォールディング モデル UniIF を提案しました。

分子の逆フォールディングは医薬品や材料の設計において重要な役割を果たしており、科学者が理想的な構造を持つ新しい分子を合成できるようになります。これまでの研究の多くは、大きな分子や小さな分子の逆フォールディングに焦点を当てており、一般的な分子の逆フォールディングについてはほとんど注目されていませんでした。
統合された一般モデルを構築するには、次の 3 つの主な課題があります。① 単位の違い:高分子は通常、タンパク質の場合はアミノ酸、RNA の場合はヌクレオチドなど、あらかじめ定義された微細構造を基本単位として使用しますが、低分子は基本単位として原子を使用します。距離、角度、テンソル積などの情報が使用されており、統一された特性評価方法が欠如しています。 ③ システム規模: 小さな分子では、グローバルな注意メカニズムが長期的な依存関係を学習できますが、これは大きな分子では機能しないことがよくあります。
上記の課題に対処し、RoseTTAFold All-Atom および AlphaFold 3 による分子構造予測の進歩をさらに補完するために、ウェストレイク大学の未来産業研究センターのチームは、すべての分子の逆フォールディングのための統一モデル UniIF を提案しました。研究者らは、UniIF の有効性を実証するために、タンパク質設計、RNA 設計、材料設計などの複数のタスクに関する包括的な実験を実施しました。結果は、UniIF がすべてのタスクで最先端のパフォーマンスを達成していることを示しています。
関連研究のタイトルは「UniIF: Unified Molecule Inverse Folding」で、トップカンファレンスNeurIPS 2024に選ばれました。
研究のハイライト:
*研究によって提案された統一モデルUniIFは、一般的な分子の逆フォールディングに対して多用途かつ効果的なソリューションを提供します
* このモデルは 2 つのレベルから統合されています。データ レベルでは、ローカル座標系の構築とモデル レベルでの幾何学的特徴の初期化、幾何学ブロック アテンション ネットワークを含む、すべての分子の統一ブロック図データ形式が提案されています。すべての分子の3D相互作用の特徴を捉えるために導入されました
* 研究者らは、提案された手法がタンパク質設計、RNA 設計、材料設計という 3 つの主要なタスクにおいて最先端の手法を上回っていることを実証しました。この成果は、機械学習、創薬、材料にプラスの影響を与える可能性があります。科学コミュニティ
用紙のアドレス:
https://arxiv.org/abs/2405.18968
公式アカウントをフォローし、バックグラウンドで「Molecular Reverse Folding」に返信すると全文のPDFが入手できます
オープンソース プロジェクト「awesome-ai4s」は、100 を超える AI4S 論文の解釈をまとめ、大規模なデータ セットとツールを提供します。
https://github.com/hyperai/awesome-ai4s
データセット: 対応するデータセットを選択して 3 つのタスク実験を実施します
タンパク質設計タスクでは、研究者は CATH4.3 データセットで UniIF を評価しています。データセットは CATH トポロジ分類コードによって分割され、その結果、トレーニング サンプルが 16,631 個、検証サンプルが 1,516 個、テスト サンプルが 1,864 個になります。
汎化能力を評価するために、研究者らは、一部のベースラインではデータ漏洩のリスクがある事前トレーニング済みの ESM2 モデルを使用していることを考慮して、時間分割戦略を採用しました。時間分割評価では、特定の日付より前のデータをトレーニング セットに割り当て、その日付以降のデータをテスト セットに割り当てます。構造の時間分割評価には、トレーニング中に見られなかった新しい結晶構造を含む CASP15 データセットが使用されました。シーケンスの時間分割評価には、2023 年 11 月 23 日より前の 30 日間が含まれる NovelPro データセットが使用されました。 76 の公開されたタンパク質配列の構造が AlphaFold 2 によって予測されました。
RNA 設計タスクでは、研究者らは、RDesign が収集した 2,218 個の RNA 三次構造を含むデータセットで RNA 実験を実施しました。データセットは、構造の類似性に基づいてトレーニング セット (1,774 個の構造)、テスト セット (223 個の構造)、および検証セットに分割されています。セット (221 構造)。データサンプルの数が少ないため、研究者らは 3 回の独立した実行からの回収率の中央値とその標準偏差を報告しています。
マテリアルデザインのタスクでは、研究者らは、単一金属酸化物由来のナノ材料マップで構成される CHILI-3K データセットで UniIF を評価しました。データセットには 53 の金属元素と 1 つの非金属元素 (酸素) が含まれており、合計 3,180 のグラフ、6,959,085 のノード、および 49,624,440 のエッジが含まれています。
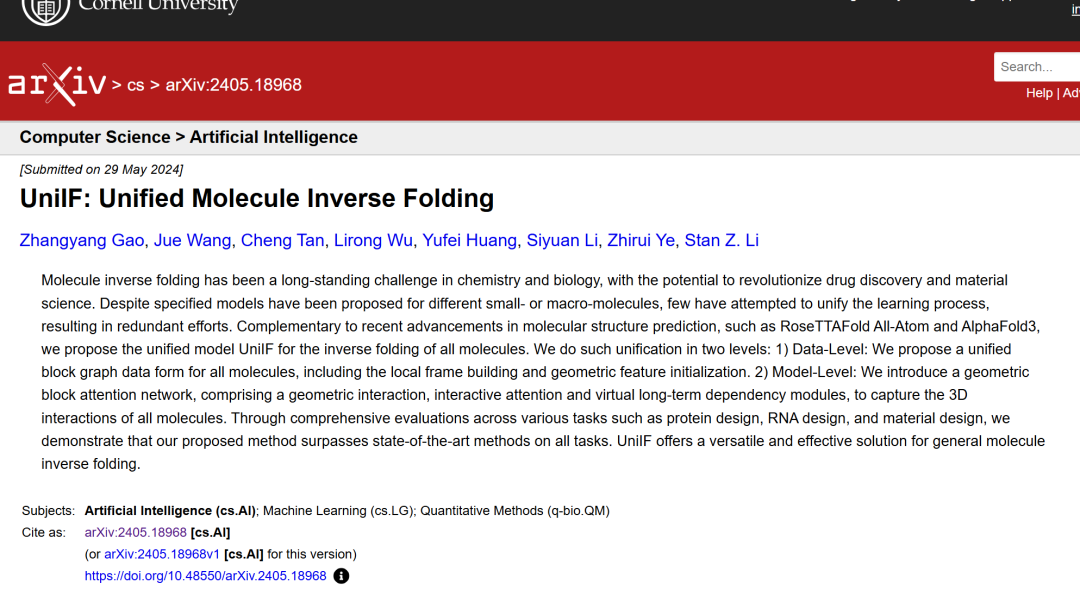
モデルアーキテクチャ: UniIF、一般的な分子逆フォールディング用の統一モデル
以下に示すように、研究者らは一般的な分子の逆折り畳みの統一モデルを提案しました。
① このモデルは、あらゆる種類の分子 (All Molecules) をブロック図に変換します。高分子 (Macromolecules) の場合は、アミノ酸とヌクレオチドに基づく事前定義されたフレームワークを使用し、低分子 (Small Molecule) の場合は、GNN ローカル フレームの層を通じて学習します。各ブロック。
② Geometric Featurizer を使用して、幾何学的ノード特徴 (Node feature) とエッジ特徴 (Edge features) を初期化します。
③ ブロック グラフ アテンション レイヤー (Block Graph Attendance) を提案し、これに基づいてブロック グラフ ニューラル ネットワーク (Block Graph Neural Network) が構築され、表現力豊かなブロック表現を学習します。
④ 最後に、UniIF がタンパク質設計、RNA 設計、材料設計などのさまざまなタスク (Task) において競争力のある結果を達成できることが実証されました。
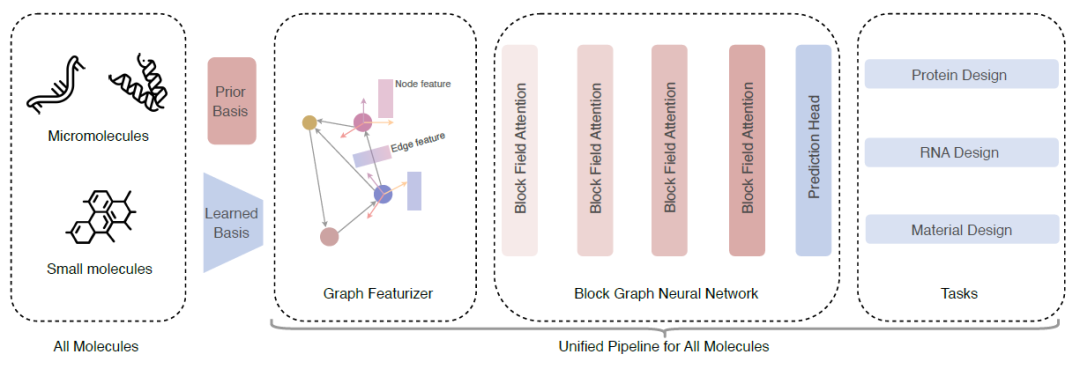
構成ブロック図:モデル アーキテクチャの最初のステップは、あらゆるタイプの分子を表すブロック グラフを導入することです。重要なのは、不規則な原子の集合 (さまざまなサイズ) を規則的なブロック表現 (固定サイズ) に変換することです。研究者らは、等変フレームと不変固有ベクトルを含むブロックと、軸行列と変位ベクトルを含むローカルフレームを使用して、すべての分子のモデリングを統一するフレームベースのブロック表現を導入しました。大きな分子の場合、軸行列はアミノ酸とヌクレオチドに基づいて事前に定義されますが、小さな分子の場合、低分子には先験的な共通の構造パターンがないため、軸行列を学習する必要があります。 n 個のブロックを含む分子が与えられたとすると、研究者らは kNN アルゴリズムを使用してブロック グラフを構築しました。
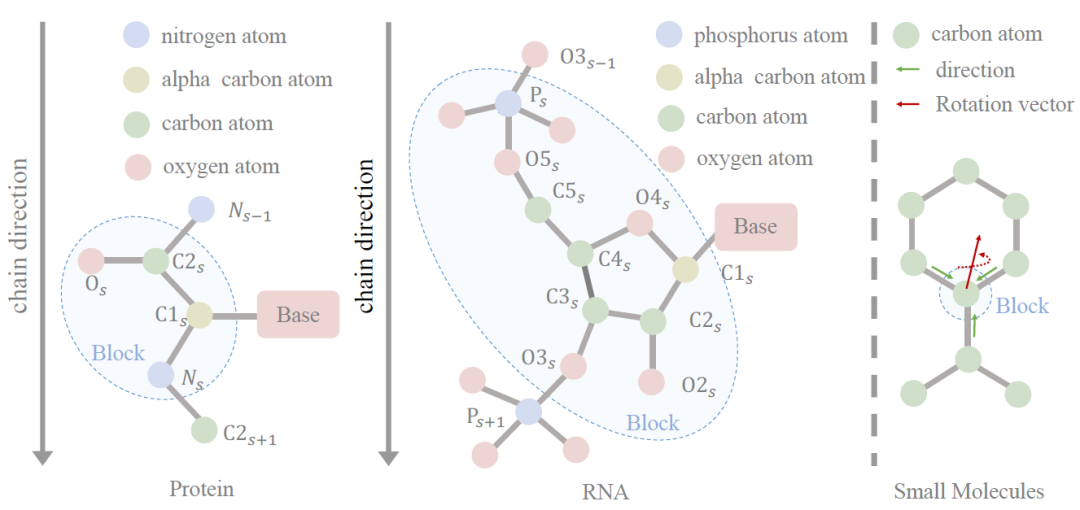
ブロックマップ特徴抽出:低分子の場合、事前定義されたローカル フレームワークは利用できないため、研究者は各原子のローカル フレームワークを学習する必要があります。つまり、分子が与えられた場合、GNN の層を使用して原子表現を初期化し、幾何学的特徴抽出器を使用して初期化します。ジオメトリ ノードとエッジ フィーチャ。
ブロックグラフアテンションモジュール:研究者らは、すべての分子の三次元相互作用を捉えるために、幾何学的相互作用、対話型注意、仮想長期依存モジュールを含む幾何学的ブロック注意ネットワークを導入しました。
調査結果: UniIF はすべてのタスクで最先端の手法を上回ります。
研究者らは、以下を含む複数の逆折り畳みタスクとアブレーション研究を通じて UniIF の有効性を実証しました。
* タンパク質設計 (T1): 標的構造に折り畳まれるタンパク質配列を設計します。
※RNA設計(T2):標的構造に折り畳まれるRNA配列を設計します。
* 材料設計 (T3): 既知の材料構造から安定した組成を発見する
①タンパク質設計(T1)
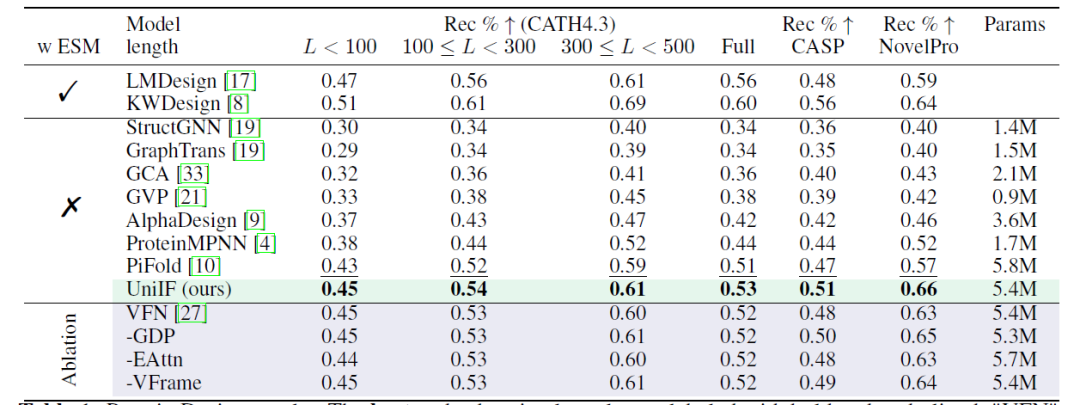
Protein Design は、標的構造に折り畳むことができるタンパク質配列を設計することを目的としており、研究者は、さまざまな設定 (ESM2 の有無にかかわらず) および複数のデータセット (CATH4.3、CASP、NovelPro) の下で結果を提供します。以下の表に示すように、ESM2 を使用せずに純粋な逆折り畳みモデルを使用すると、UniIF はすべてのデータセットで最高のパフォーマンスを達成し、その有効性を実証します。
*LMDesign と KWDesign には ESM2 が含まれますが、StructGNN、GraphTrans、GCA、GVP、AlphaDesign、ProteinMPNN、PiFold には ESM2 は含まれません。
CATH4.3 では、強力なベースライン モデルのため、全体的な改善は限られていますが、時分割評価では、UniIF の汎化能力における利点が、学習可能なパラメータが少ないにもかかわらず、強力なベースライン PiFold を上回っていることが強調されています。時間分割評価では、UniIF は ESM2 ベースの手法を含むすべてのベースラインを大幅に上回っています。新しいシーケンスを含む NovelPro では、UniIF はシーケンス最適化に ESM2 を使用して LMDesign および KWDesign よりも優れたパフォーマンスを発揮します -これは、UniIF が実用的なアプリケーションにとって重要な優れた汎化能力を備えていることを示しています。
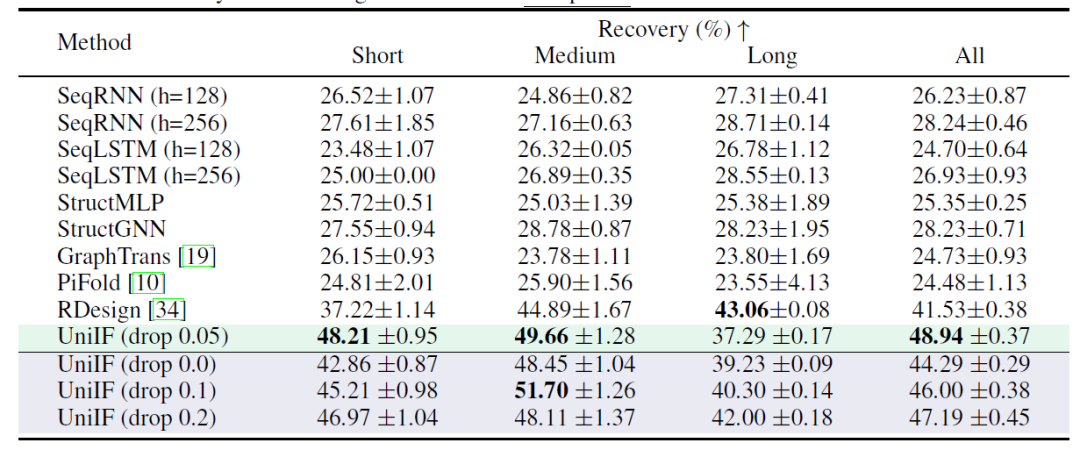
②RNA設計(T2)
RNA 設計の目標は、標的構造に折り畳まれる RNA 配列を設計することです。以下の表に示されているように、UniIF はすべてのケースで最高のパフォーマンスを達成します。これは、PiFold などの以前の強力なベースライン モデルがタンパク質設計でのみ良好なパフォーマンスを示していたため、大幅な改善です。報告されているのは、UniIF は、タンパク質と RNA の設計タスクの両方で最先端のパフォーマンスを達成した最初のモデルであり、その多用途性と有効性を実証しています。
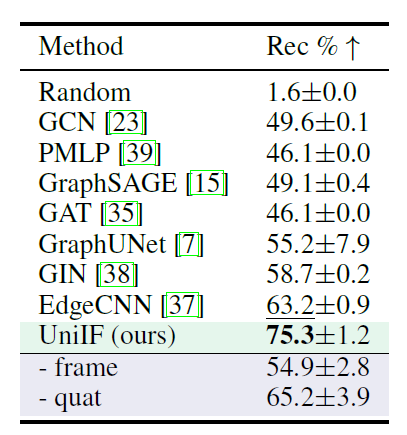
③マテリアルデザイン(T3)
既知の材料構造から安定した原子の組み合わせを発見することは新材料の発見にとって重要であるため、研究者らはこの新しいタスクにおけるUniIFのパフォーマンスも評価した。以下の表に示すように、UniIF は、すべてのベースライン モデルを大幅に上回ります。
④事例紹介
下の画像では、研究者らが設計したタンパク質とRNAの配列を示しています。さらに、AlphaFold 3 を使用して、設計された配列を構造にリフォールディングしました。実際の構造 (灰色)、PiFold 構造 (緑色)、および UniIF 構造 (ピンク) を並べて比較しました。研究者らは次のように観察しました。UniIF は回復率と二乗平均平方根偏差 (RMSD) の両方の改善を達成し、逆折りたたみタスクにおけるその有効性を示しています。

UniIF モデルは AlphaFold 3 をさらに補完します
一般的な分子学習は近年ますます注目を集めており、RoseTTAFold All-Atom (RFAA) と AlphaFold 3 はこの方向で大きな成功を収めた 2 つの代表的なモデルです。
2024 年 3 月 7 日、David Baker は、「RoseTTAFold All-Atom を使用した一般化された生体分子モデリングと設計」というタイトルの研究論文を Science 誌に発表しました。研究チームは、アミノ酸と DNA 塩基の残基ベースの表現と他のすべてのグループの原子表現を組み合わせた RoseTTAFold All-Atom (RFAA) を開発しました。これにより、タンパク質、核酸、小分子、金属、配列と化学構造の分析が可能になります。共有結合的に修飾されたコンポーネントがモデル化されます。
原紙:
https://www.science.org/doi/10.1126/science.adl2528
2024 年 5 月 9 日、Demis Hassabis 氏、John Jumpe 氏らは「AlphaFold 3 との生体分子相互作用の正確な構造予測」と題する研究論文を Nature 誌に発表しました。この研究では、リガンド (低分子)、タンパク質、核酸 (DNA と RNA) がどのように集合し、相互作用するのかなど、タンパク質データ バンクのほぼすべての分子タイプを含む複合体の構造を予測できる最新モデルである AlphaFold 3 を発表しました。これらの分子システムに対する翻訳後修飾やイオンの構造的影響を予測することにより、研究者が生体分子システムの構造を原子レベルで正確に観察するのに役立ちます。
原紙:
https://www.nature.com/articles/s41586-024-07487-w
2 つのモデルを詳しく見てみると、RFAA は小さな分子を表すために原子結合図を使用し、大きな分子を表すためにフレームワーク図を使用します。AlphaFold 3 は、すべてのモデルに適用できる 2 層表現、つまり原子表現とラベル表現を使用します。分子。タグの概念は、アミノ酸やヌクレオチドなどの原子のグループを表す、前述のブロックの概念と同等です。
GET と EPT は、小分子と大分子の両方に適したブロック表現を採用し、新しい等変トランスフォーマー スケルトンを導入する、最近提案された 2 つのモデルです。低分子の原子結合図を指定する RFAA とは異なり、この記事で紹介する UniIF モデルは、すべての分子タイプに統一されたブロック図を採用しており、原子結合図を必要としません。さらに、このモデルでは、それぞれにベクトル基底も導入されています。ブロック。これは AlphaFold 3 と一致します。GET と EPT は異なります。
普遍的な分子モデルを構築するという課題はある程度解決されたので、UniIF モデルは、RoseTTAFold All-Atom や AlphaFold 3 などの「先駆者」の分子構造予測の方向性における進歩をさらに補完するものとみなすことができます。将来的には、継続的に反復される大規模生物学モデルは、研究者が生物学の世界を再理解し、創薬を再考するのに役立ち、それによって全人類に利益をもたらすでしょう。
参考文献:
1.https://arxiv.org/abs/2405.18968
2.https://mp.weixin.qq.com/s/8OvxVlUuZZZ2gcepIl5UBw
3.https://www.jiqizhixin.com/articles/2024-03-08-6
4.https://m.thepaper.cn/newsDetail_forward_28984037









