Command Palette
Search for a command to run...
動的なタンパク質のドッキング予測を実現!上海交通大学/星耀科技/中山大学などが共同で幾何学深度生成モデルDynamicBindを立ち上げ

タンパク質は生命の物質的基盤であり、その機能はタンパク質の構造や立体構造の動態と密接に関係しており、リガンドによって制御されています。タンパク質とリガンドの相互作用の研究は、創薬やスクリーニングにとって非常に重要です。研究プロセス全体を通じて、AlphaFold の発売は画期的な進歩であり、単一タンパク質の空間三次元構造を予測することができ、タンパク質-リガンド相互作用を研究するための構造的基礎を提供します。
しかし、AlphaFold はタンパク質の静的な構造を瞬間的に予測することしかできず、タンパク質の構造の動的な変化を予測することはできません。AlphaFold によって予測されたリガンドのないタンパク質構造がドッキングの入力として使用される場合、結果として得られるリガンド位置の予測は、多くの場合、リガンド結合共結晶構造と一致しません。さらに、AlphaFold によって予測される構造は、リガンドとの結合に最適な側鎖および主鎖の配置を示すことが難しく、関連する活性部位が正しい位置にないため、AlphaFold の構造を使用することは現時点では困難です。薬物のスクリーニングと設計に使用されます。
これを考慮して、上海交通大学のZheng Shuangjia氏の研究グループは、Xingyao Technology、中山大学薬学部、米国のライス大学と協力して、タンパク質の動的ドッキング用に設計された幾何学的深さ生成モデルDynamicBindを提案した。これは、タンパク質の立体構造を初期の AlphaFold 予測状態からホロ様の状態に効果的に調整することができ、深層学習に基づく新しい研究パラダイムを提供し、AlphaFold 後の時代の創薬のためのタンパク質の動的変化を考慮します。この方法は、国際医薬品スクリーニング競争 CACHE における湿式実験でも検証されており、パーキンソン病治療の薬剤化が困難な標的に対して競合可能性のある化合物をスクリーニングすることができます。
この研究は「DynamicBind: 深層等変生成モデルによるリガンド特異的タンパク質-リガンド複合体構造の予測」と題され、Nature Communications に掲載されました。
研究のハイライト:
* 高度な深部拡散モデルと等変幾何学ニューラル ネットワーク技術を使用して、タンパク質立体構造生成とリガンド位置予測を 1 つのフレームワークに統合し、タンパク質とリガンドの動的なドッキング予測を実現します。
* DynamicBind は、タンパク質とリガンドのドッキングにおける従来のドッキング手法や深層学習に基づくリジッドドッキング手法よりも優れています。
* DynamicBind は、AlphaFold によって予測されたタンパク質の立体構造を使用して、タンパク質の立体構造を動的に調整し、リガンドに最も適した最適な立体構造を見つけます。

用紙のアドレス:
https://www.nature.com/articles/s41467-024-45461-2
オープンソース プロジェクト「awesome-ai4s」は、100 を超える AI4S 論文の解釈をまとめ、大規模なデータ セットとツールを提供します。
https://github.com/hyperai/awesome-ai4s
データ セット: PDBbind データ セットに基づいて、MDT テスト セットを使用して評価範囲を拡張します。
研究者らはまず、実験的に測定された結合親和性と組み合わせた PDBbind データセットを使用して、トレーニング、検証、テスト セットで時系列順にモデルをトレーニングしました。PDBbind テスト セットには、多くの非低分子リガンド (53 個はペプチド) を含む約 300 の 2019 構造が含まれているため、研究者らは厳選された主要薬物標的 (MDT) テスト セットを使用して評価の範囲を拡大しました。
MDT テスト セットには、2020 年以降にアーカイブされた 599 個の構造が含まれており、これにはキナーゼ、GPCR、核内受容体、イオン チャネルを含む 4 つの主要なファミリーの薬物様リガンドとタンパク質が含まれます。これらのタンパク質ファミリーは、FDA が承認した小分子薬の約 70% 標的を表しており、ある程度代表的です。
DynamicBind: 幾何学深層学習に基づく動的化合物構造予測モデル
タンパク質をほとんど剛体として扱う従来のドッキング手法とは異なり、DynamicBind は、高度な深部拡散モデルと等変幾何学ニューラル ネットワーク テクノロジーを使用して、タンパク質の立体構造生成とリガンド位置の予測という従来分離されていた 2 つのステップを 1 つのフレームワークで統合し、タンパク質の動的ドッキング予測とリガンドが実現します。同時に、エンドツーエンドの深層学習手法として、広範囲のタンパク質の立体構造変化のサンプリングにおいて、従来の MD シミュレーションよりも数桁高速です。
DynamicBind は、構造様 (apo 様) PDB 形式と、SMILES 形式や SDF 形式などの広く使用されているいくつかの小分子リガンド形式を受け入れます。推論中、モデルはリガンドをランダムに配置し、RDKit を使用してタンパク質の周囲にリガンドのシード構造が生成されます。トレーニング段階では、モデルはアポ様構造からホロ構造へのプロセスを学習することを目的としています。推論中、モデルは初期入力構造を 20 回繰り返し更新します。
以下の図 a に示すように、ピンクはタンパク質の立体構造のホログラフィック状態 (ホロ) を表し、緑色は初期のアポリポタンパク質とモデルの予測立体構造を表し、シアンは天然のリガンドを表し、オレンジは予測されたリガンドを表します。
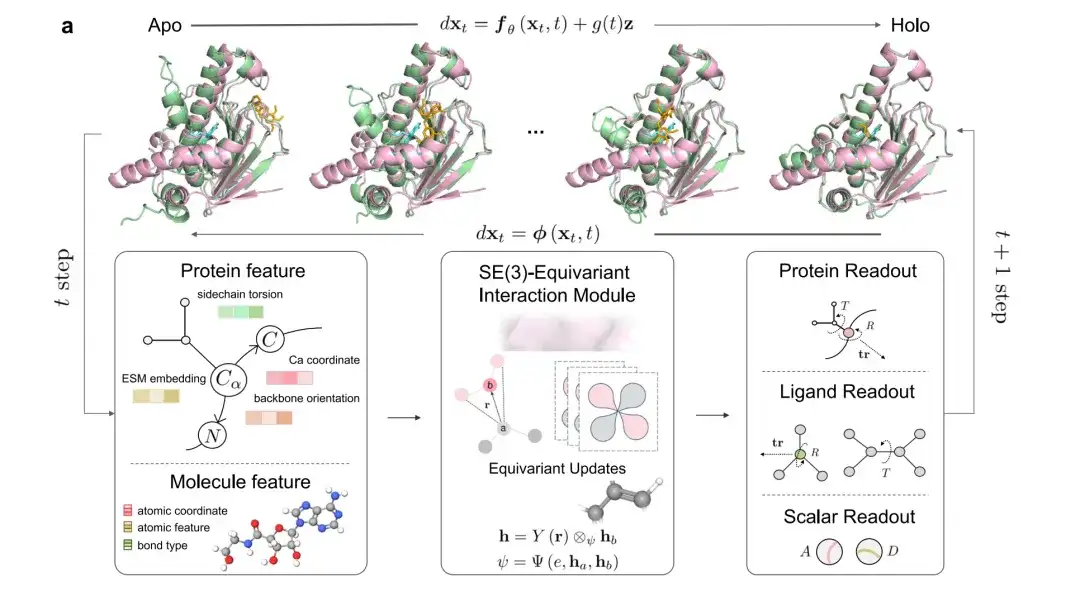
各反復で、タンパク質とリガンドの特性と座標 (側鎖のねじれ、Ca 原子座標など) が SE(3) Equivariant Interaction Module に入力されます。モデルの出力結果には、リガンドと各タンパク質残基の全体的な平行移動と回転、リガンドのねじれ角とタンパク質残基のカイ角の回転、および 2 つの予測モジュール (結合親和性 A と信頼スコア D) が含まれます。
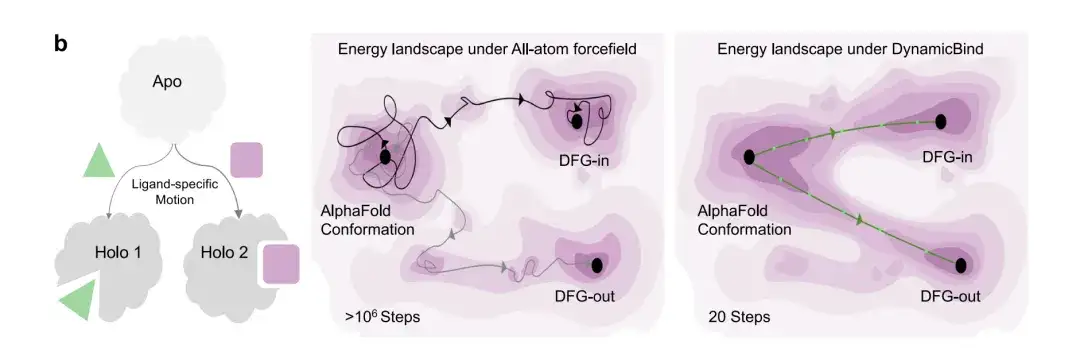
以下の図 b は、DynamicBind モデルのサンプリング効率を示しています。キナーゼタンパク質の DFG-in から DFG-out への遷移中に、タンパク質が 2 つの異なるリガンドに結合する場合、このモデルは 2 つの異なるホログラフィック立体構造を予測できます。また、DynamicBind は結合タンパク質の立体構造を 20 ステップで予測できますが、同じ結合状態を探す全原子 MD シミュレーションには数百万ステップが必要です。
タンパク質の動的ドッキング予測の万能ツールである DynamicBind は、5 つの主要なタスクで優れたパフォーマンスを発揮します。
DynamicBind のモデルのパフォーマンスを評価するために、研究者らは次の 5 つの主要なタスクを通じてそれをテストしました。
(1) DynamicBind と現在のドッキング方法のベンチマーク テスト。
(2) 多数のタンパク質の構造変化をサンプリングする能力。
(3) タンパク質の立体構造変化の範囲に対処する。
(4) 動的ドッキングを達成するための不可解なポケットの能力を予測します。
(5) 抗生物質ベンチマークにおけるスクリーニング性能。
DynamicBind は、従来のドッキング手法や深層学習に基づくリジッド ドッキング手法よりも優れています。
テスト中、研究者らはホログラフィック構造を入力として使用せず、ホログラフィックタンパク質立体構造は利用できないと想定し、AlphaFold によって予測されたタンパク質立体構造のみを入力として使用しました。ホログラフィック構造は、共結晶化したリガンドとの強い形状および電荷相補性を示すため、リガンドの姿勢予測のプロセスが簡素化されます。
以下の図 a および b に示すように、研究者らは、PDBbind データ セットおよび MDT データ セットで DynamicBind を他のベースライン モデルと比較しました。異なる RMSD しきい値の下では、DynamicBind が他の方法よりも優れたパフォーマンスを示しました。具体的には、RMSD 閾値が 2Å (5Å) 未満の DynamicBind リガンドの割合は、PDBbind テスト セットでは 33% (65%) であり、MDT テスト セットでは 39% (68%) です。
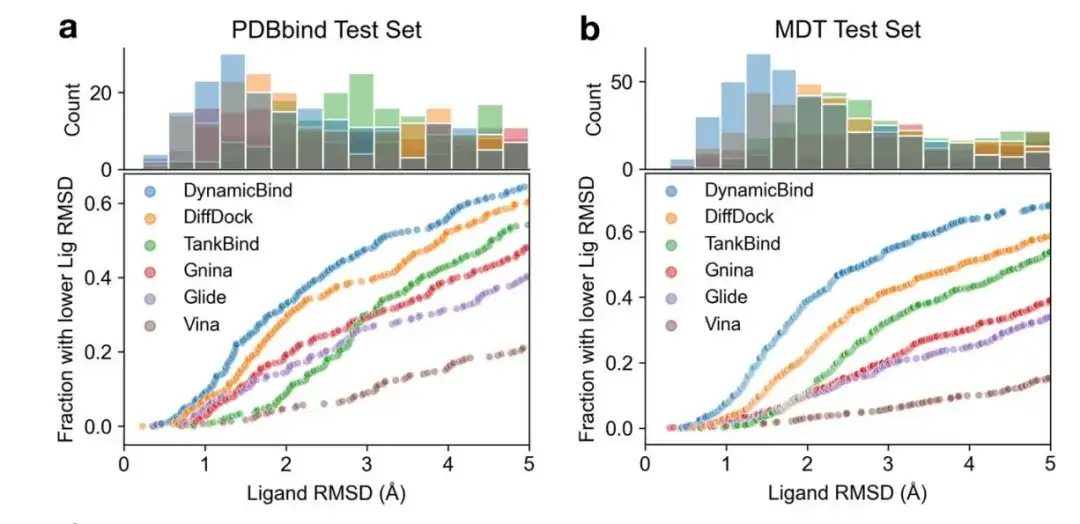
ただし、モデルを評価する場合、DiffDock、TankBind、DynamicBind などの深層学習ベースのモデルは、Van を厳密に実装する場合に有利です。デル ワールス力 (ファン デル ワールス力)、力場ベースのドッキング手法である Gnina、Glide、Vina は不利であり、モデル評価の客観性に影響を与えます。したがって、研究者らはリガンド RMSD と競合スコアを使用してリガンド予測の成功率を評価しました。
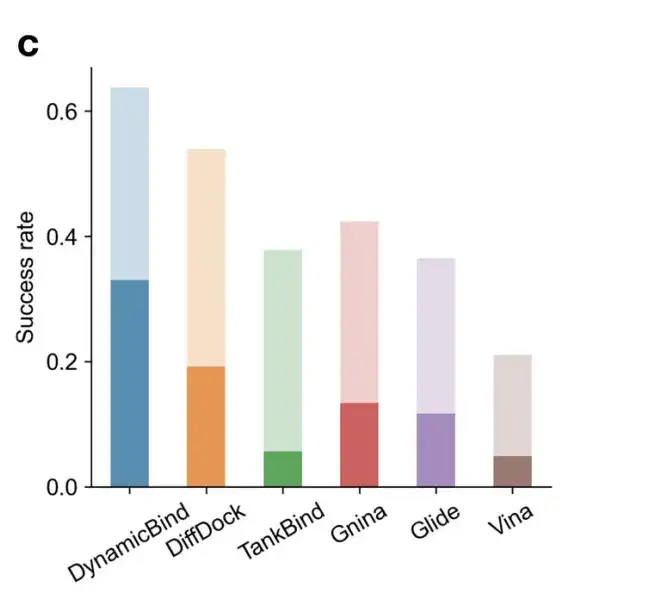
以下のパネル c は、厳密な基準 (リガンド RMSD < 2 Å、競合スコア < 0.35) とより緩和な基準 (リガンド RMSD < 5 Å、競合スコア < 0.5) を使用したリガンド予測の成功率を示しています。より厳しい条件下では、DynamicBind の成功率 (0.33) は、最良のベースライン DiffDock (0.19) より 1.7 倍高くなります。
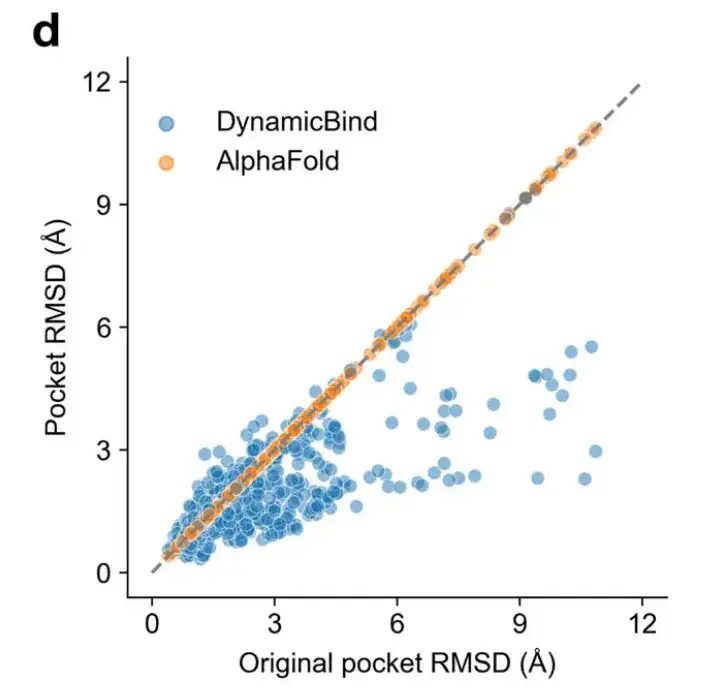
さらに、初期ポケットと結晶構造の間の RMSD が大きい場合でも、以下の図 d に示すように、DynamicBind によって予測されたポケット RMSD は AlphaFold によって予測された結果よりも大幅に小さくなります。
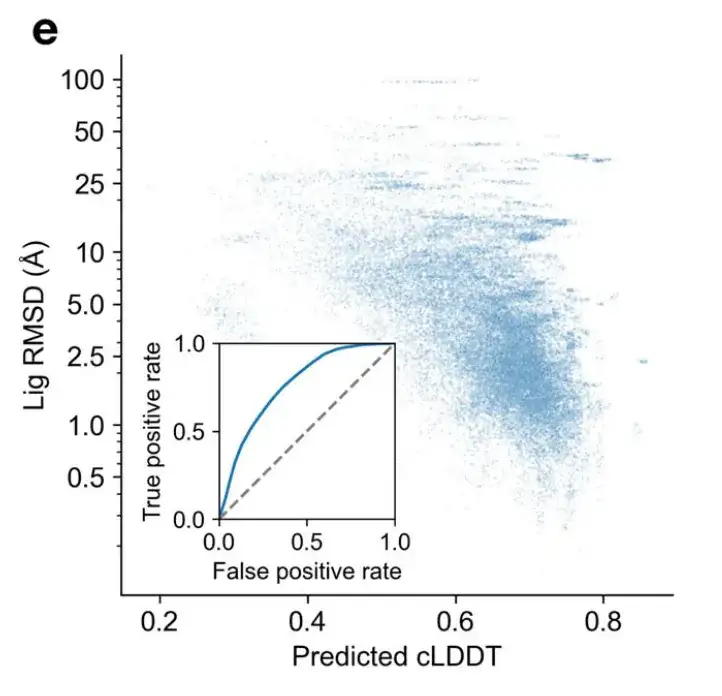
DynamicBind が多様な立体構造を生成できることを考慮し、AlphaFold の LDDT スコアに触発されて、研究者らは予測出力から最適な複雑な構造を選択するための contact-LDDT (cLDDT) スコアリング モジュールを開発しました。
以下の図 e に示すように、DynamicBind によって予測された cLDDT は実際のリガンド RMSD とよく相関しており、高品質の複雑な構造の選択におけるその有効性を示しています。
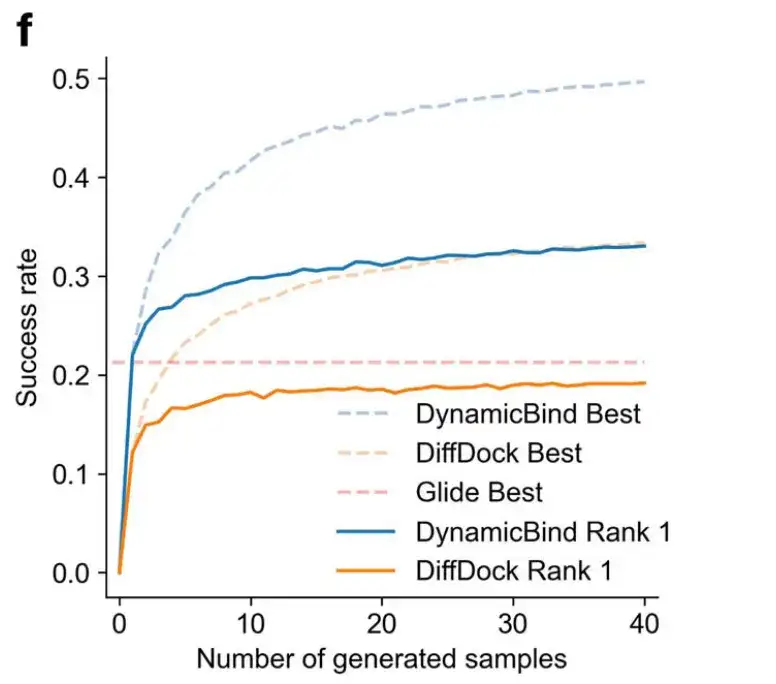
以下の図 f に示すように、生成されるサンプルの数が増加するにつれて、リガンドのポーズを予測する DynamicBind モデルの成功率も増加します。
DynamicBind はリガンド特異的なタンパク質の構造変化を捕捉します
従来のドッキングプロトコルでは、通常、タンパク質の立体構造サンプリングがドッキングプロセスとは別のステップとして実行されます。ただし、多くの場合、2 つの異なるリガンドが相互に排他的なタンパク質の立体構造に適している可能性があります。以前のドッキング モデルでは、リガンドの適切な結合姿勢を特定する前に、タンパク質を正しい立体構造に事前設定する必要がありました。対照的に、DynamicBind は、AlphaFold によって予測されたタンパク質の立体構造を使用して、タンパク質の立体構造を動的に調整し、目的のリガンドに最も適した最適な立体構造を見つけます。以下の図 a を参照してください。
図bからeは、PDB 6UBWおよびPDB 7V3Sの構造におけるDynamicBindおよびAlphaFoldの予測リガンドおよびポケットのRMSDを示しています。 PDB 6UBW の場合、DynamicBind はリガンド RMSD が 0.49 Å、ポケット RMSD が 1.97 Å であると予測しましたが、AlphaFold 構造のポケット RMSD は 9.44 Å でした。 PDB 7V3S の場合、DynamicBind はリガンド RMSD が 0.51 Å、ポケット RMSD が 1.19 Å (AlphaFold 6.02 Å) であると予測しました。
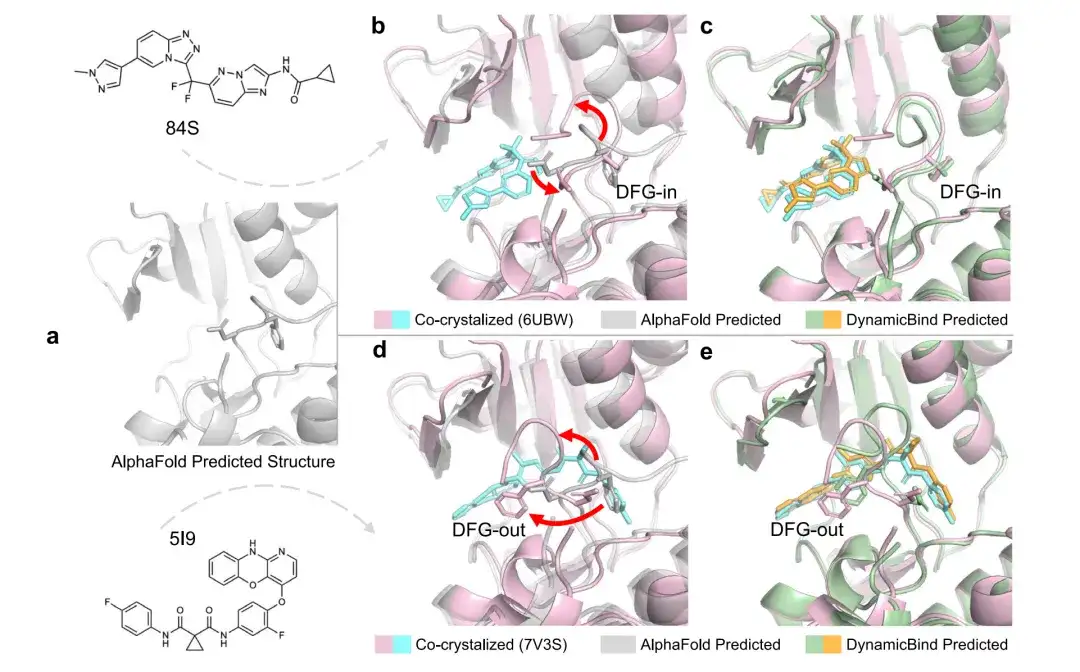
図 f と g は、UniProt ID でタグ付けされたタンパク質が、同じ初期構造から開始して、I 型阻害剤に結合すると徐々に DFG-in 立体構造に向かって移動し、II 型阻害剤と相互作用すると DFG-in 立体構造に向かう傾向を示しています。 DFG-out コンフォメーションのプロセス。
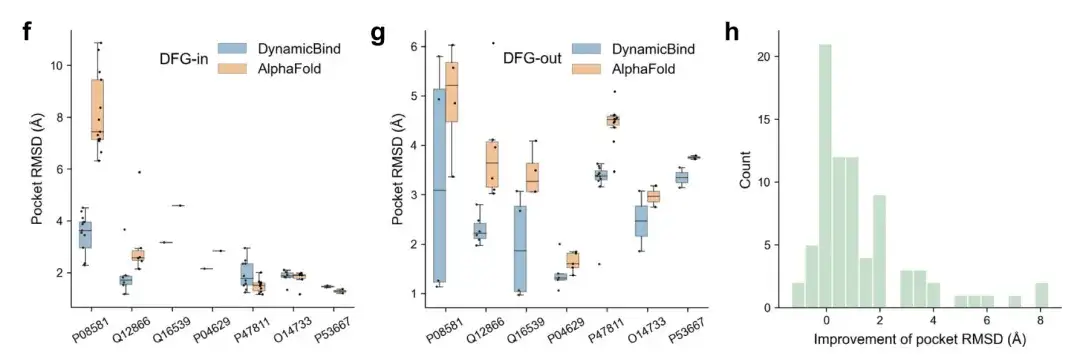
図 h は、DynamicBind から予測されたタンパク質構造のほとんどが、初期の AlphaFold 構造と比較してポケット RMSD が低いことを示しています。
上記の結果は、DynamicBind がリガンド特異的な構造変化を捕捉できることを示しています。つまり、特定の立体構造が最初に提供されたタンパク質構造と異なる場合でも、DynamicBind は、タンパク質の他の可能な立体構造によく結合する化合物を特定できます。
DynamicBind は複数のスケールでタンパク質の構造変化をカバーします
研究者らは、ピコ秒からミリ秒までのスケールにわたる 6 つの異なるタイプの構造変化を使用して DynamicBind を評価しました。以下の図に示すように、ピンクは結晶構造を表し、白は AlphaFold 構造を表し、緑は DynamicBind によって予測された構造を表し、シアンはネイティブのリガンドを表し、オレンジは DynamicBind によって予測されたリガンドを表します。
結晶構造との比較から、Δポケット RMSD は、モデルで予測されたタンパク質構造と AlphaFold 構造の間のポケット RMSD の差を測定します。負の Δpocket RMSD は、DynamicBind によって予測された構造が、結晶構造よりも AlphaFold によって予測された構造に近いことを示します。
Δclash は、AlphaFold 構造内の予測されたタンパク質-リガンドのペアとグラフトされたリガンドの間の競合スコアの差を測定します。負の Δclash は、予測された複合体内の衝突が少ないことを示します。
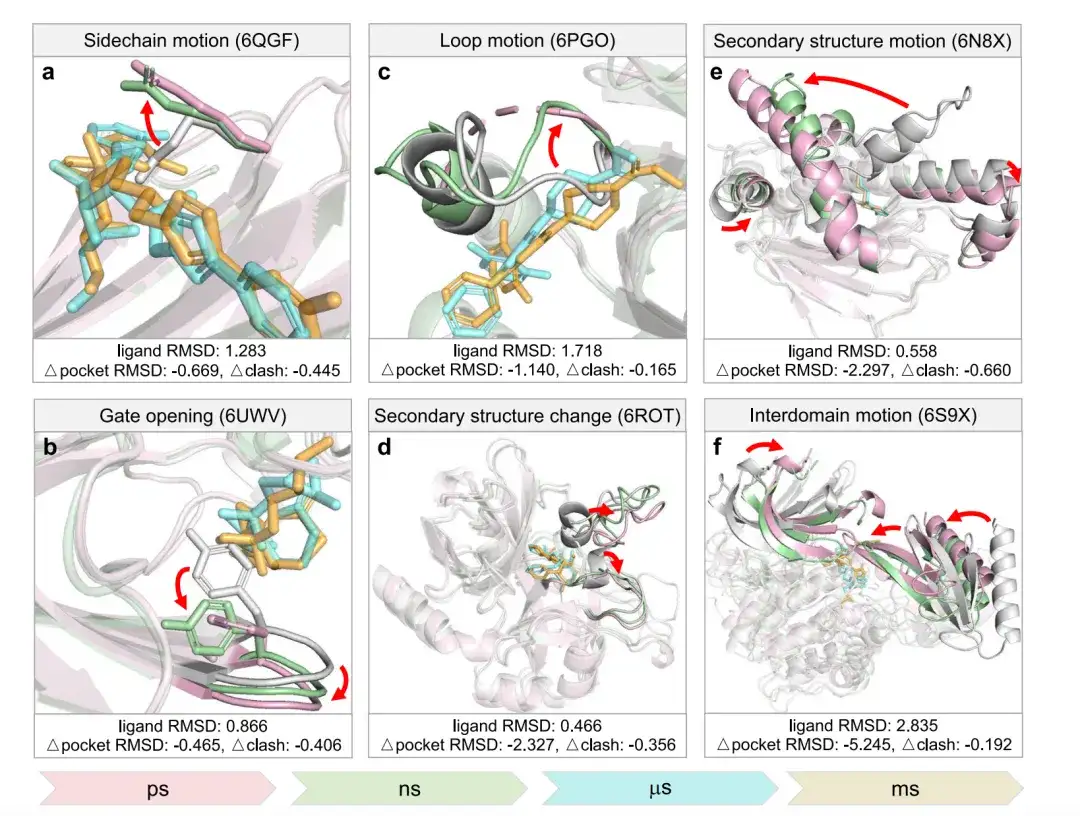
図aでは、ネイティブのリガンドが重ね合わされたAlphaFold構造の側鎖と衝突しますが、DynamicBindの予測では、この側鎖はネイティブの立体構造に向かって移動し、それによって衝突が解決されます。図 b では、AlphaFold 構造のチロシンがポケットの一部をブロックしますが、DynamicBind の予測構造とネイティブ構造では、ポケットのこの部分がアクセス可能になります。パネル c では、柔軟なループがリガンドと交差していますが、DynamicBind の予測では、天然の構造と一致して離れています。
パネル d では、α ヘリックスがリガンド結合部位の近くでループになります。図eでは、熱ショックタンパク質Hsp90αが閉じた状態から開いた状態に遷移する際に、二次構造が大きく動きます。パネル f では、AKT1 キナーゼの 2 つのドメインが結合して、以前には存在しなかったポケットを形成します。
要約すると、DynamicBind モデルは、リガンド結合ポケットの広さが十分でない場合、または AlphaFold によって予測される立体構造を形成しない場合に、リガンド結合に関連するさまざまな立体構造変化を予測できます。
DynamicBind は不可解な結合サイトを特定します
タンパク質の動的プロセスは隠れたポケットを作成することが多く、静的な構造では見つからない薬効部位が明らかになる可能性があり、これまで「創薬不可能」だったタンパク質が潜在的な創薬標的となる可能性があります。研究者らは、ケーススタディとして SET ドメイン含有タンパク質 2 (SEtD2) を使用して、これらの隠れたポケットを明らかにする際の DynamicBind の有用性を実証しています。
多発性骨髄腫 (MM) およびびまん性大細胞型 B 細胞リンパ腫 (DLBCL) の治療における重要な薬剤であるヒストン メチルトランスフェラーゼである SETD2 には秘密のポケットがあり、選択性の高い化合物 EZM0414 です。この標的は現在第 I 相臨床試験中です。 。
以下の図aおよびbに示すように、トレーニングセット内のすべてのSETD2ホモログ(0.4を超えるタンパク質のスミス-ウォーターマン類似度に従って定義)は、S-アデノシルメチオニン(SAM)またはシネファンギン類似体と共結晶化されました。シアン色のバーは PDB 7TY2 のリガンド EZM0414 を表し、ピンク色のバーはタンパク質を表します。
パネル c では、白は AlphaFold 構造とその表面を表しており、隠れた部位がブロックされており、移植された EZM0414 と多数の競合が生じています。
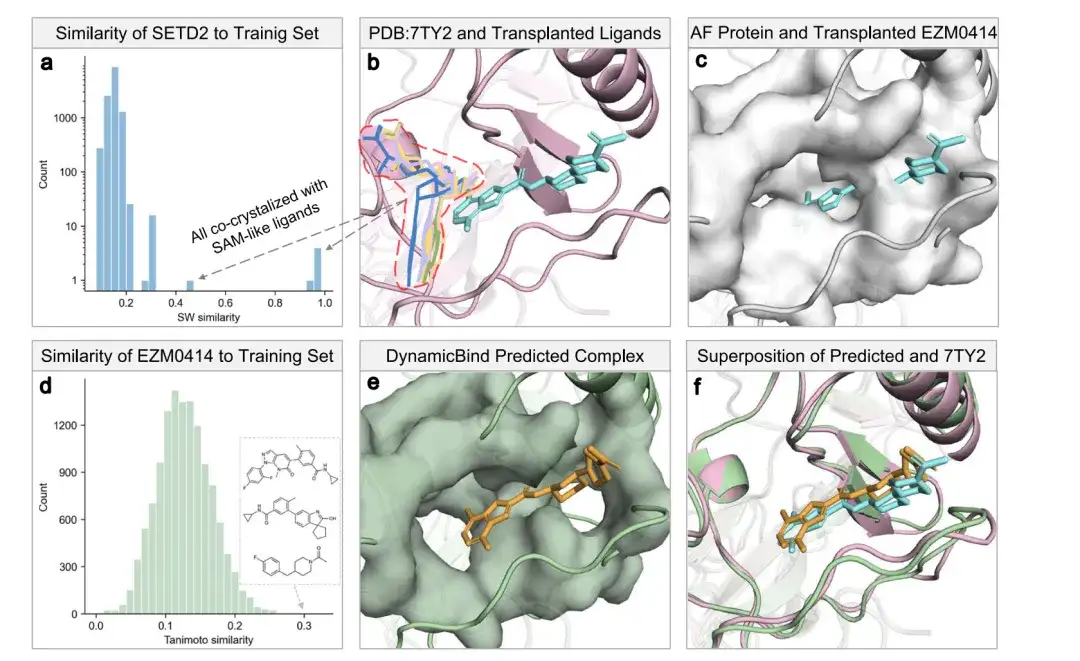
図 d は、EZM0414 が目に見えないリガンドであり、最も類似したタニモト リガンドでさえ EZM0414 から大きく逸脱していることを確認しています。パネル e は、AlphaFold によって予測された SETD2 構造および EZM0414 の SMILES 表現を入力として受け取る DynamicBind モデルによって予測されたタンパク質-リガンド複合体構造を示しています。図 f は、DynamicBind によって予測されたタンパク質-リガンド複合体の構造と SETD2-EZM0414 複合体の結晶構造 (PDB 7TY2) の間の重複を示しています。
結果から判断すると、DynamicBind は隠れたポケットの動的ドッキングを達成し、リガンドの配置に成功しただけでなく、より適切なポケット構造も見つけました (結果として得られるリガンド RMSD は 1.4 Å、ポケット RMSD は 2.16 Å)。
DynamicBind により、抗生物質ベンチマークにおける薬物スクリーニング パフォーマンスの向上が可能になります
ターゲットベースの創薬プロセスでは、潜在的な薬剤候補のスクリーニングと逆スクリーニング(特定の化合物のタンパク質ターゲットの同定)の両方が重要です。DynamicBind モデルのスクリーニング性能を実際に評価するために、研究者らはモデルに親和性予測モジュールを追加し、PDBbind データセットから得られた実験的に測定された結合親和性データを使用してトレーニングし、2023 年に Drug Proteome of Antibiotics に発表しました。 スクリーニング テストの評価データ (12 のタンパク質ターゲットと 3,000 近くの測定された活性データを含む)。
以下の図 a に示すように、DynamicBind は、VINA や DOCK6.9 などの一般的なドッキング方法や、機械学習に基づく最良の再スコアリング方法を上回り、受信機動作特性曲線下平均面積 (auROC) は 0.68 に達しました。このパフォーマンスの向上は、DynamicBind の動的ドッキング機能によるもので、AlphaFold 構造をネイティブ状態に近づけることができ、より正確な結合親和性の推定が可能になります。
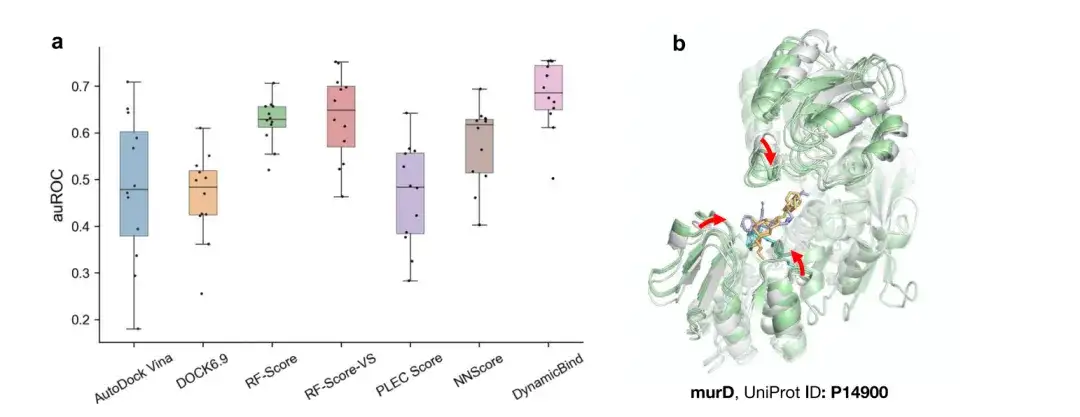
上のパネル b は、DynamicBind によって予測されたタンパク質 murD の構造がリガンドをより緊密に取り囲み、初期の AlphaFold 構造では不可能だったより多くの相互作用を形成していることを示しています。
これらの結果は、DynamicBind が従来のドッキング手法や深層学習ベースのリジッドドッキング手法よりも一貫して優れていることを示しており、このモデルはその結合親和性予測機能により、プロテオームレベルでの仮想スクリーニングアプリケーションにおいて大きな可能性を示しています。
タンパク質の複雑な構造と機能を解読し、インテリジェントな創薬に貢献する
AlphaFold の静的構造予測に基づいて、DynamicBind モデルは生成人工知能テクノロジーを革新的に導入し、動的化合物構造予測の課題を解決します。タンパク質構造の動的な変化を予測することは、生命プロセスの理解や新薬の開発にとって非常に重要です。特にAI医薬品の研究開発において、AI医薬品スクリーニングの精度と臨床有用性を大幅に向上させることができます。
この研究の主要な参加者の一人として、鄭双嘉氏の研究チームは、生成人工知能と医薬品開発の交差点における最先端の研究に長年従事し、実りある成果を上げてきました。
2024 年 6 月 21 日、Zheng Shuangjia の研究グループは、表現型レベルで摂動されたハイコンテンツの細胞顕微鏡画像を使用して分子表現の学習を支援するクロスモーダル学習方法を提案しました。この方法は、分子と特性評価の間のギャップを効果的に埋めることができ、これは医薬品開発にとって非常に重要です。関連する研究のタイトルは「細胞画像を使用したクロスモーダル グラフの対照学習」で、『Advanced Science』誌に掲載されました。
用紙のアドレス:
https://onlinelibrary.wiley.com/doi/10.1002/advs.202404845
2024 年 5 月 25 日、Zheng Shuangjia 氏の研究グループは、原子構造スケールと分子ネットワーク スケール間のマルチスケール情報を効果的に統合するマルチスケール学習フレームワーク MUSE を提案しました。計算による創薬を他のスケールに拡張する可能性を実証します。関連する研究は「タンパク質と薬物の相互作用のバランスのとれたマルチスケール学習のための変分期待値最大化フレームワーク」と題され、Nature Communications に掲載されました。
用紙のアドレス:
https://www.nature.com/articles/s41467-024-48801-4
2022 年 9 月 15 日、Zheng Shuangjia 氏の研究グループは、創薬が困難な標的向けの生成的インテリジェント創薬アルゴリズムを開発し、短期間で PROTAC リード化合物を設計しました。動物実験でも実証されており、情報技術とバイオテクノロジーの融合の大きな可能性が実証されています。この一連の結果は、Google の DeepMind AlphaFold チームやワシントン大学の学者 David Baker チームなど、この分野のトップの研究グループによって積極的に引用され、評価されています。関連する研究のタイトルは「深層学習と分子シミュレーションによる合理的な PROTAC 設計の加速」で、Nature Machine Intelligent に掲載されました。
用紙のアドレス:
https://www.nature.com/articles/s42256-022-00527-y
2020 年 2 月 14 日、Zheng Shuangjia の研究グループは、エンドツーエンドの深層学習フレームワークに基づく準視覚的な質問応答システムを提案しました。薬物とタンパク質間の相互作用を特定し、創薬を効果的に支援します。関連する研究は「準視覚的質問応答システムを使用した薬物とタンパク質の相互作用の予測」というタイトルで、Nature Machine Intelligence に掲載されました。
用紙のアドレス:
https://www.nature.com/articles/s42256-020-0152-y
Zheng Shuangjia 氏の研究グループは、生成人工知能と医薬品開発の交差点における最先端研究の理解に基づいて、代謝と老化に関連する疾患に対する医薬品のインテリジェントな設計に焦点を当て、IT と BT を統合した新しい医薬品開発モデルを作成しています。 、エンドツーエンドのスマートドラッグを提供することに尽力しています。貢献の力を発見してください。
参考文献:








