Command Palette
Search for a command to run...
AI for Genomics | 空間トランスクリプトームデータ表現アルゴリズム SPACE、ゲノミクスにおける人工知能アプリケーション

生放送の「Meet AI4S」シリーズの 2 回目のエピソードでは、幸運なことに、清華大学生命科学部の Zhang Qiangfeng 研究室の博士研究員である Li Yuzhe 氏を招待します。張強峰氏の研究室は清華大学生命科学部に属しており、清華大学・北京大学共同生命科学センターおよび北京先端構造生物学革新センターの重要な一部でもある。同研究室の研究は、生命科学と人工知能アルゴリズムの交差点、RNA構造群技術とアルゴリズム開発、単細胞ゲノム配列決定技術とアルゴリズム開発、クライオ電子顕微鏡データに基づくタンパク質構造モデリング、および関連する人工知能の開発に焦点を当てている。インテリジェンスアルゴリズムは待ってください。
この共有、Li Yuzhe 博士は、「ゲノミクスにおける AI アプリケーションの探索: 空間トランスクリプトーム データ表現アルゴリズム SPACE を例として」というテーマで講演しました。チームの最新の研究結果を共有し、空間トランスクリプトミクスおよび単一細胞オミクス研究における AI 手法を紹介しました。
HyperAI Super Neural は、Li Yuzhe 博士の詳細な共有を、当初の意図に違反することなく編集し、要約しました。
クリックして完全なライブ リプレイを表示します。
AI for Science は科学分野の研究パラダイムに大きな変化をもたらします
今日は、AI for Science が科学分野全体の研究パラダイムに大きな変化をもたらしたと思います。次に、タンパク質の構造の研究を例に挙げて詳しく説明します。
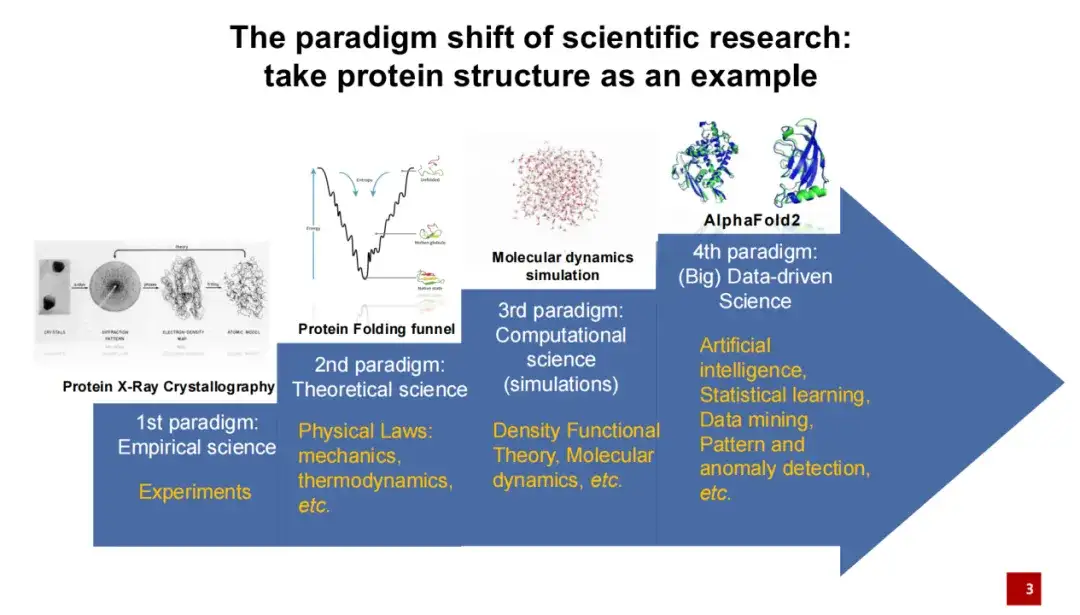
第一世代のタンパク質構造研究パラダイムは、主に実験的手段を通じて行われました。つまり、タンパク質が形成する結晶をX線で撮影し、構造モデリングを行います。
第 2 世代のタンパク質構造研究パラダイムには、主にタンパク質構造の研究に理論的知識を追加する一部の物理学者が関与しています。たとえば、タンパク質が低エネルギーで折りたたまれる場合、この折りたたみは比較的安定しています。
第 3 世代のタンパク質構造研究パラダイムは 1990 年代を指し、コンピューター技術の発展に伴い、コンピューター シミュレーションがタンパク質構造研究に徐々に適用されました。特に、分子動力学シミュレーションは近年広く利用されている。これらのシミュレーション手法は、タンパク質の構造をある程度正確に計算し、予測するのに役立ちます。近年、特に 2020 年には、人工知能アルゴリズムがタンパク質構造の分野に参入し、新たなブレークスルーをもたらしました。 2020 年のタンパク質構造予測コンテストでは、AlphaFold 2 は他の競合手法と比べてはるかにリードしています。
人工知能の導入は、生命科学と科学研究分野全体に大きなパラダイムシフトをもたらしました。従来の調査方法と比較して、人工知能は、データから出発し、データ駆動型の科学研究を実施することに重点を置いています。これは、事前に科学的仮説を立てる必要がなくなり、その代わりにデータから直接自然法則を学び明らかにすることを意味します。
ゲノミクスAI開発の歴史
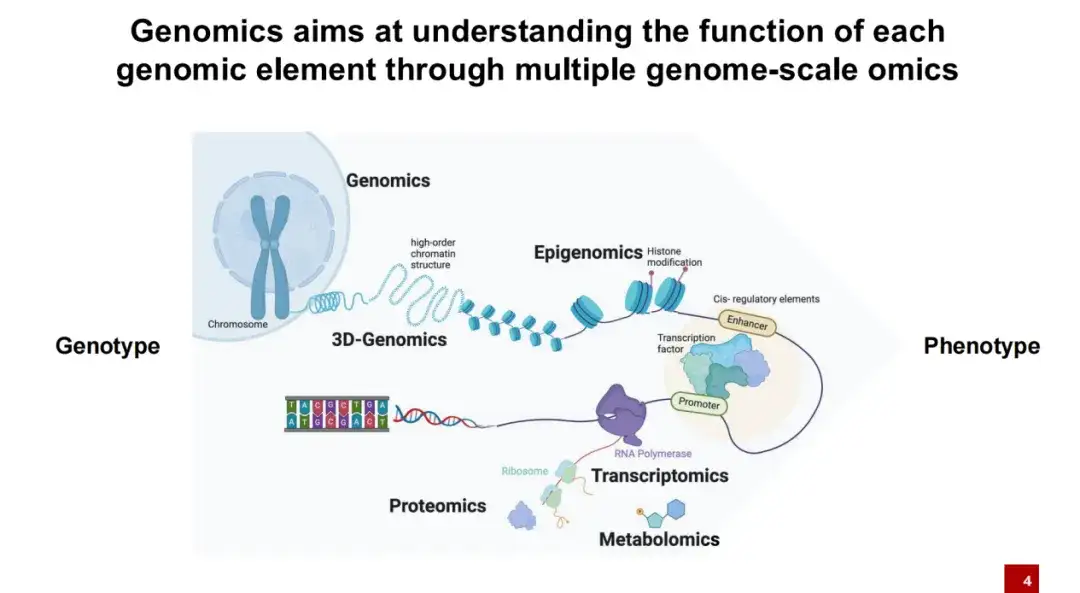
以下の共有は主にゲノミクス分野における AI の応用に焦点を当てています。要するに、ゲノミクス研究は主に、遺伝子型 (遺伝子型、つまり体内のすべての DNA) と表現型 (身長、体重、その他の個人の特徴などの表現型) の関係を調査します。
誰もが知っているように、DNA は細胞内に裸で存在するのではなく、ヌクレオソームに包まれて存在します。通常、ヌクレオソームには多くのヒストン修飾が結合しており、これらの DNA は一定の条件下でのみ露出して隙間を形成します。このとき、転写因子などのタンパク質は、これらの露出した DNA 間隔に結合することができます。
その後の転写プロセスでは、RNA は RNA ポリメラーゼによって転写され、その後リボソームによってタンパク質に翻訳され、最終的にタンパク質は生命活動の中で機能します。ゲノミクスの研究目標は、さまざまな DNA 要素が生命活動にどのような影響を与えるかを理解することです。
1950年代にDNAの二重らせん構造が解読されて以来、近年の科学用AIの開発における重要な出来事と開発プロセスをまとめました。その始まりは、1950 年代の DNA の二重らせん構造の発見と 1970 年代のサンガー配列決定技術の開発にまで遡ります。
以下の図に示すように、青色の部分はさまざまなシーケンス技術と実験技術の開発を表し、緑色の部分はいくつかの重要な大規模研究計画とデータベースの構築を表しています。 ; 赤い部分は、ゲノミクス分野における代表的な手法とアプリケーションを表す AI です。
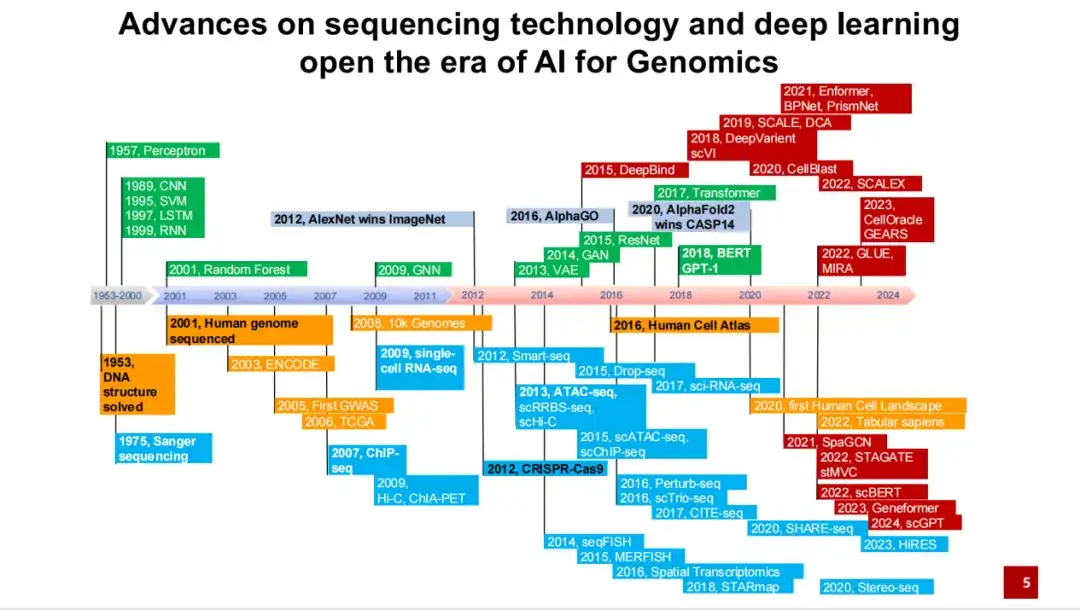
ご覧のように、2001年、ヒトゲノム計画の草案は当初完成し、白人男性の全DNA配列が解読された。 2012年、画像分類タスクにおける AlexNet のパフォーマンスは初めて人間を超え、人工知能の分野で 10 年以上にわたる爆発的な発展の先駆けとなりました。 2016年、Human Cell Atlas Project が提案され、研究は徐々に単一個人の DNA 配列からすべての細胞に移行しました。同年、強化学習法に基づくAlphaGoが囲碁ゲームで人間を破った。
ゲノミクスAIやサイエンスAIの分野では、重要な進歩は、AlphaFold 2 が 2020 年の CASP 14 で大差の 1 位を獲得したことです。これにより、ゲノミクスの分野で人工知能手法がますます適用されるようになりました。
で、単細胞ゲノミクスは、近年のゲノミクス分野における大きな進歩です。従来のゲノミクス研究では通常、バルクシーケンスが実行されます。以下の図の各線は細胞の種類を表し、異なる色の線は異なる細胞の種類を表すと想定されています。これまでの配列決定方法では、組織全体に対して混合配列決定を実行していたため、各 DNA または RNA がどの特定の細胞に由来するかを判断することが困難でした。単一細胞技術の出現により、組織内のすべての DNA または RNA を取得できるだけでなく、これらの DNA または RNA の特定の細胞起源を特定することも可能になります。細胞の種類が異なれば遺伝子発現も異なり、機能も異なるため、生命活動をさらに理解することができます。
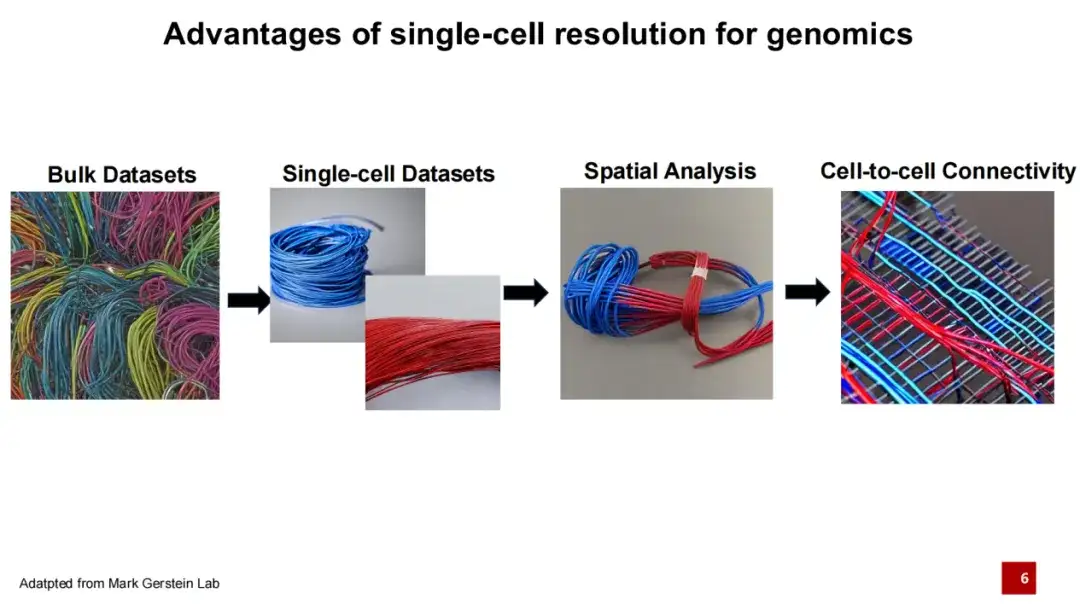
過去 5 年間で、空間トランスクリプトームに代表される空間オミックス技術は、単一細胞オミックス技術に基づいてさらに一歩前進しました。各細胞タイプに関する情報を取得できるだけでなく、空間内でのこれらの細胞の分布も決定できます。細胞間の相互作用はそれらの機能を実現するための重要な基盤であるため、細胞がどのように接続されているかに焦点を当ててさらなる研究が行われています。
ヒトゲノム プロジェクトの開始以来、2016 年にヒト セル アトラス プロジェクトが提案されるまで、その目標は、生命活動をより深く理解し、特定の疾患の治療と診断をサポートするために、すべてのヒト細胞の参照マップを完成させることでした。 。
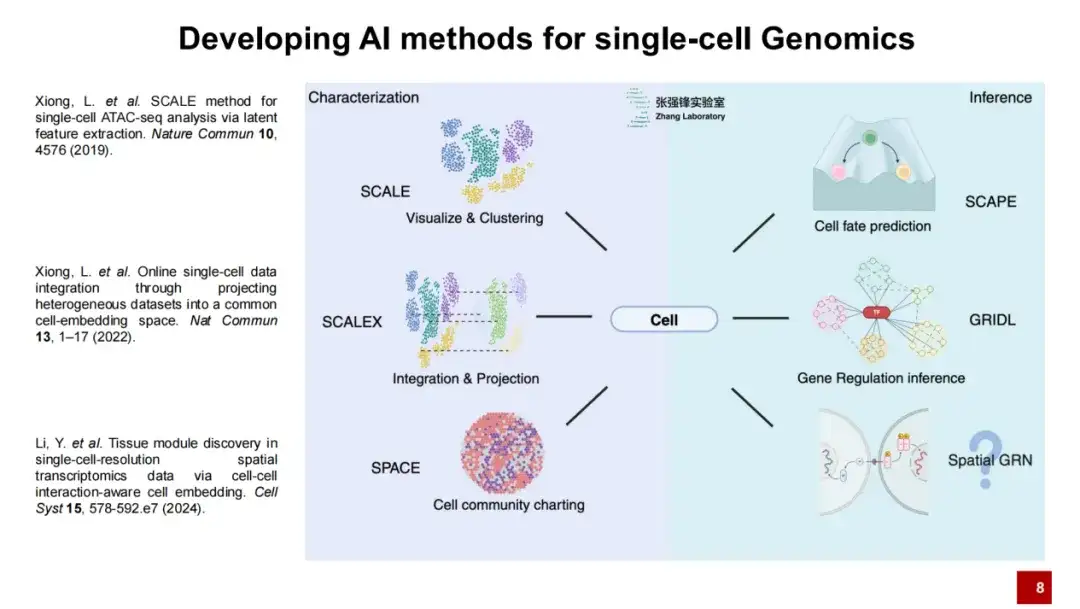
研究チームは、単一細胞ゲノミクス研究を実施するために、SCALE、SCALEX、SPACE という 3 つの手法を開発しました。
私たちの研究室は一連の人工知能手法を開発してきました。私たちは、単一細胞ゲノミクスには 2 つの主要なステップが必要であると考えています。1 つ目は細胞の説明、2 つ目は細胞の推論です。
現在、細胞を描いた作品としてSCALE、SCALEX、SPACEの3作品を出版しています。SCALE は主に視覚化とクラスタリングに関するもので、SCALEX はデータの統合と投影に関するもので、SPACE は空間トランスクリプトーム データ構成全体の微小環境の記述に関するものです。今回は主にSCALEXとSPACEの2つの方法を紹介します。
バッチ効果を排除する SCALEX メソッド
SCALEX の手法は、バッチ効果 (バッチ効果) を排除することです。これはゲノミクス研究において非常に重要な問題です。バッチ効果とは、異なる実験条件やその他の技術的要因による、異なるバッチの実験結果の差異を指します。
下の図に示すように、2 セットの生物学的に複製する細胞を別々に培養したとしても、これら 2 セットの細胞の配列を決定すると、理論的には非常に類似した遺伝子発現が得られるはずです。しかし、培養環境の違い、ライブラリ構築時間の違い、シーケンスプラットフォームの違いなどの技術的な理由により、最終的な遺伝子発現プロファイルには大きな違いがあり、それによって大量の技術的なノイズが発生する可能性があります。したがって、データを分析する際には、バッチ効果のこの部分を削除する必要があります。

生物学の研究では、データは一度に収集できないことが多く、実験を重ねることで徐々に蓄積されます。したがって、バッチ効果を除去し、データを統合して、本当に生物学的に関連する因子を見つけます。これは、ゲノミクスまたは単一細胞ゲノミクス研究における重要なステップです。
これをもとにSCALEX工法を開発し、処理された単一細胞データを一般化された細胞潜在空間に投影できます。 SCALEX のフレームワークは、変分オートエンコーダー (VAE) に基づいています。
最初の入力は単一細胞のトランスクリプトーム データであり、バッチフリー エンコーダーを通じて一般化された細胞の潜在空間に投影されます。
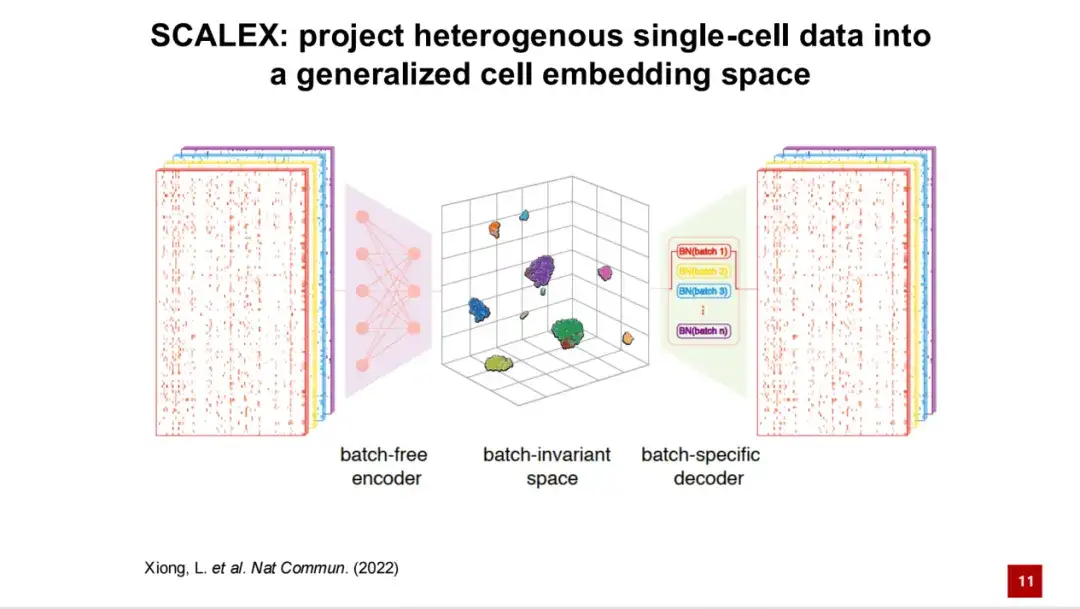
次に、バッチ固有のデコーダによるドメイン固有のバッチ正規化を通じて、バッチ情報がモデルに追加されます。この非対称設計により、生成されたセルの潜在空間はバッチに依存しない空間となり、理論的にはバッチ関連の技術的ノイズが含まれません。遺伝子発現はデコーダーを通じて再構築され、元の入力遺伝子発現プロファイルを使用して損失 (Loss) が計算され、KL 発散と結合されて SCALEX モデルの損失関数 (Loss Function) が形成されます。監修モデル。
この非対称エンコーダおよびデコーダ設計には、次の 2 つの主な利点があります。まず、得られたエンコーダはユニバーサルであり、つまり、モデルを再トレーニングしたり、新しいデータを既存のデータに再統合したりする必要がなく、エンコーダーを介したバッチ情報なしで、新しいデータをセルの潜在空間に直接投影できます。
第二に、SCALEX はバッチ全体の効果により多くの注意を払っています。バッチ効果を除去する従来の方法は、主に 2 つのデータ バッチ内で類似したセル (セル ペア) を見つけてそれらをペアにしてバッチ効果を除去することです。この方法は、本質的には比較的局所的なバッチ効果です。
ただし、このタイプの方法の問題は、実際のデータ分析では、2 つの異なるバッチのセル タイプが完全に一致しない可能性があり、共通するセル タイプはわずかで、残りはバッチ固有である可能性があることです。セルのペアを強制的に検索すると、適切なペアのセルが見つからずに過剰補正が発生したり、揃えるべきでない種類のセルが強制的に揃えられてしまう可能性があります。
そこで、SCALEXの2つの大きなメリットについて、さらに詳しくご説明させていただきます。
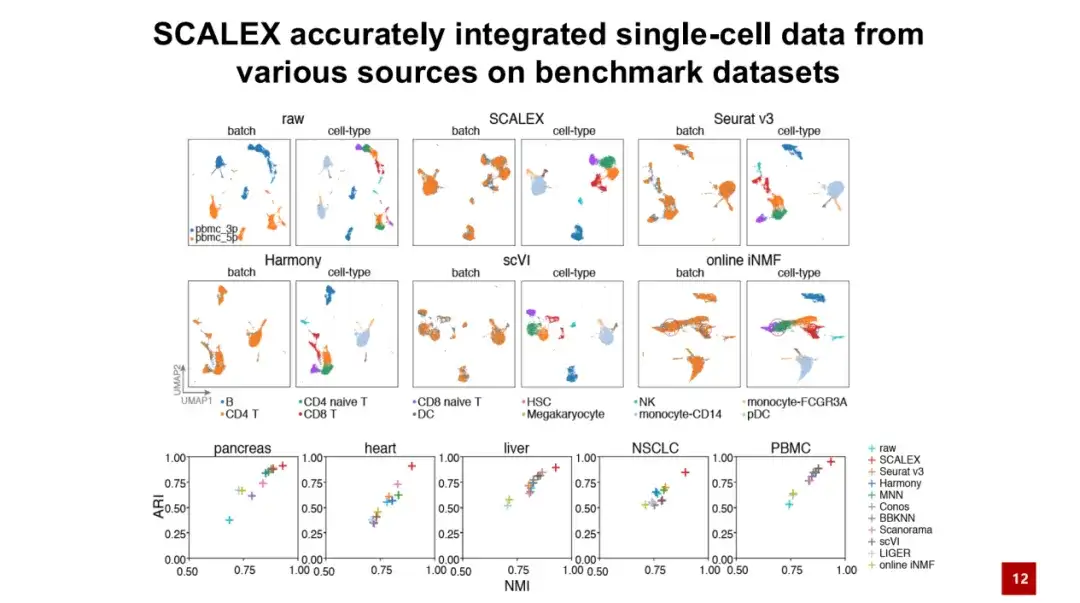
まず、5 つのテスト データ セットで SCALEX のベンチマークを実行しました。結果は、SCALEX が精度の点で既存の方法よりも優れていることを示しています。
以下の図に示すように、バッチ チャートは元のデータと未修正のデータを表し、青とオレンジはそれぞれ 2 つのバッチのデータを表し、cell-type はセルのタイプを表します。この 2 つのバッチには類似した細胞型が存在しますが、過剰なバッチ効果により、本来同じ細胞型に属する細胞が凝集できず、その結果、技術的要因が生物学的要因を覆い隠してしまい、その後の解析ができないことがわかります。 . 生物学的研究。
SCALEX を介して統合した後、2 つのセルのバッチは十分に凝集し、細胞の種類に応じて明確に分離され、実用的なアプリケーションにおける SCALEX の重要性が実証されました。
SCALEX の重要な利点は、同じ種類のセルを含む 2 つのバッチのデータを処理できることです。この種のデータは、部分重複データセットと呼ばれます。以下の図に示すように、オーバーラップ 0 は 2 つのバッチのセル タイプが完全に異なることを意味し、オーバーラップ 4 は 2 つのバッチに 4 つの共有セル タイプがあることを意味します。
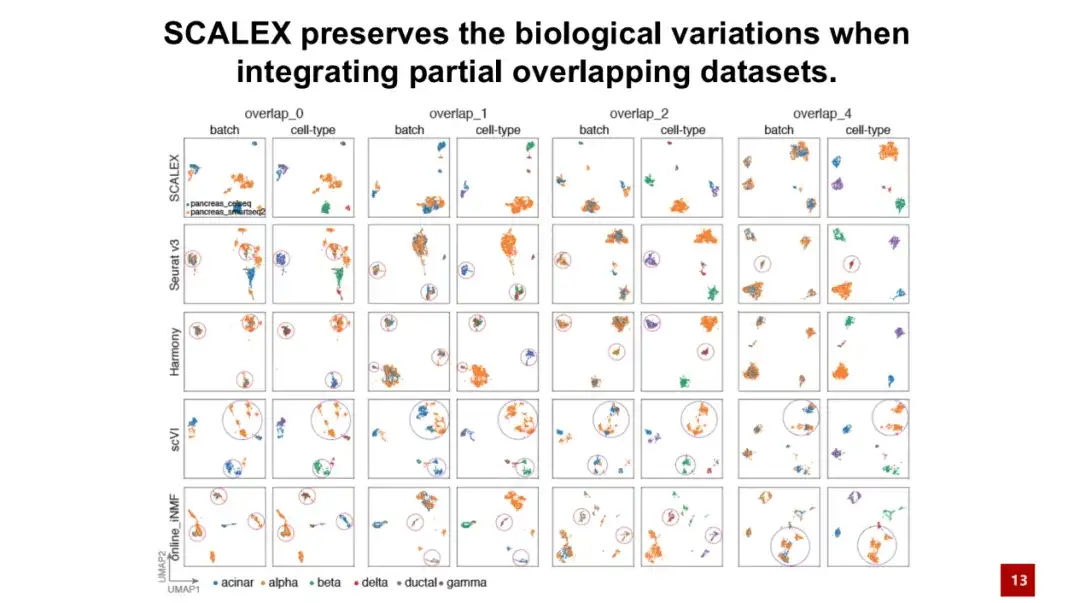
結果は、SCALEX が、2 つの細胞バッチに同じ細胞型がまったく含まれていない場合でも、生物学的差異を良好に維持していることを示しています。つまり、SCALEX は、他の同様の方法に依存する場合のように、異なる細胞型の細胞を強制的に統合しません。セルペアを見つけて過剰補正を引き起こします。
SCALEX のもう 1 つの利点は、ユニバーサル エンコーダーが、モデルを再トレーニングすることなく、新しいデータを既存のバッチフリー セル潜在空間に直接投影できることです。以下の図に示すように、参照細胞アトラスは最初に膵臓データセットを通じてトレーニングされ、次に 3 つの新しいデータがトレーニングされたエンコーダーを通じて細胞潜在空間に直接投影されます。図の色はセルのタイプを表し、灰色の点は構築された参照セルのタイプを表します。異なる細胞タイプが図のそれぞれの位置で十分に分離されていることがわかります。
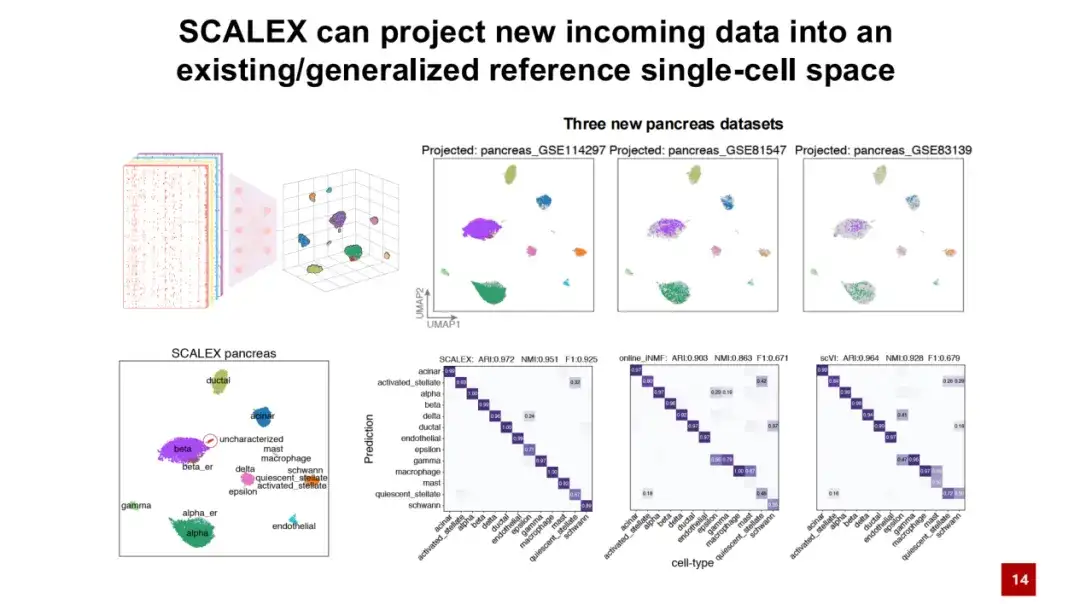
SCALEX は、特定の位置の周囲の参照セルから新しいデータ セルにマーカーを投影することにより、セル タイプに自動的に注釈を付けることに優れていることがわかります。他の既存の手法と比較して、SCALEX は非常に重要なアプリケーションを示します。つまり、新しいデータを構築されたデータに直接投影して、データ間の比較分析を行うことができます。
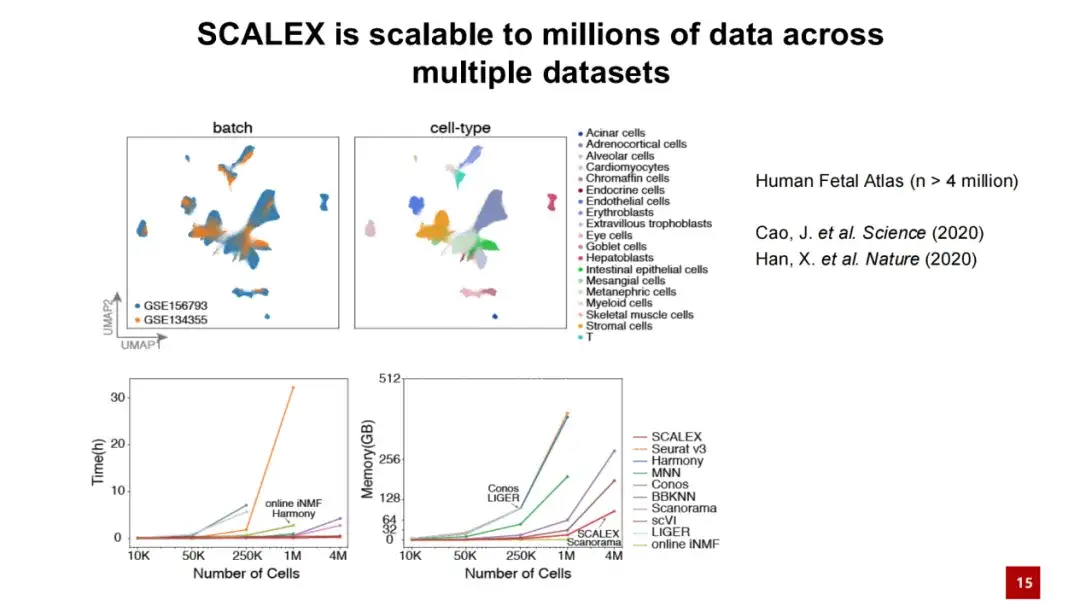
さらに、SCALEX は大規模なデータを処理する場合にも優れたパフォーマンスを発揮します。以下の図は、SCALEX が 400 万セル データを処理する場合、計算時間は数十分を超えず、メモリ消費量は 100 GB 未満であることを示しています。これは、SCALEX が優れた拡張性を備えており、非常に大規模な単一細胞データの統合分析に使用できることを示しています。
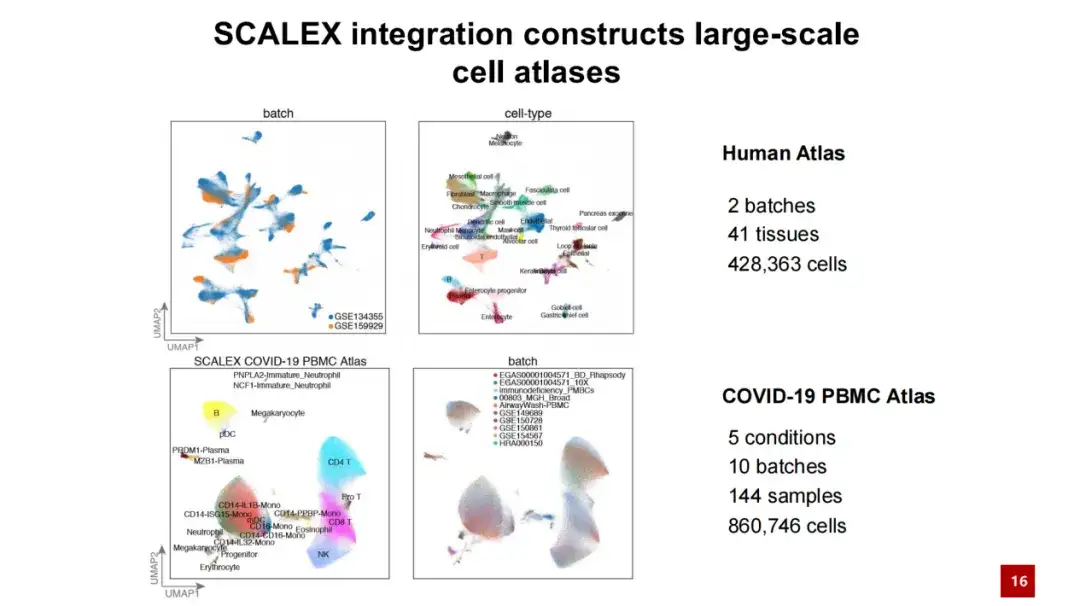
SCALEX を利用して、2 つの大規模な細胞アトラスを構築しました。1 つは、400,000 個を超える細胞を含む、ヒト個人に関する細胞アトラスで、もう 1 つは、860,000 個を超える細胞と 100 の複数のサンプルを含む、COVID-19 PBMC 細胞アトラスです。
空間トランスクリプトームデータ人工知能解析ツール SPACE
次に、チームが最近公開した空間トランスクリプトーム解析ツール SPACE を紹介します。
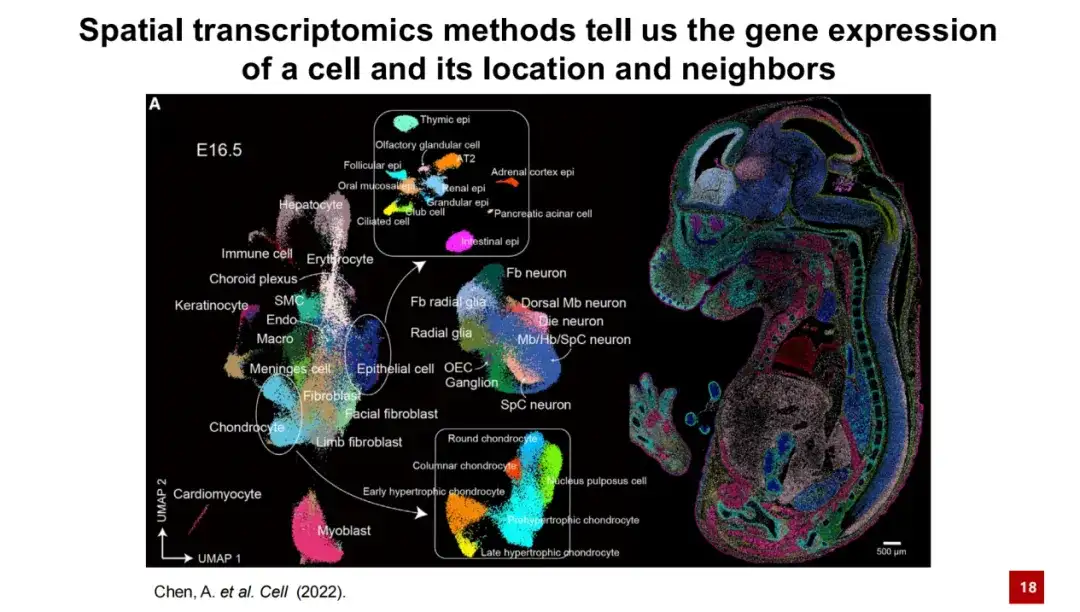
簡単に言えば、空間トランスクリプトーム技術は、細胞遺伝子発現情報とその空間内の特定の位置を提供できます。以下の図は、典型的な空間トランスクリプトームの結果を示しています。左側の画像では、各ドットはセルを表し、色はそのセルの種類を示します。これらのセルは、遺伝子発現の次元削減クラスタリングを通じて UMAP グラフを形成します。右側のパネルには、マウス胚 E16.5 データ内の各細胞の実際の空間位置が示されています。細胞が良好な特異性をもって空間的に分布していることがはっきりとわかります。
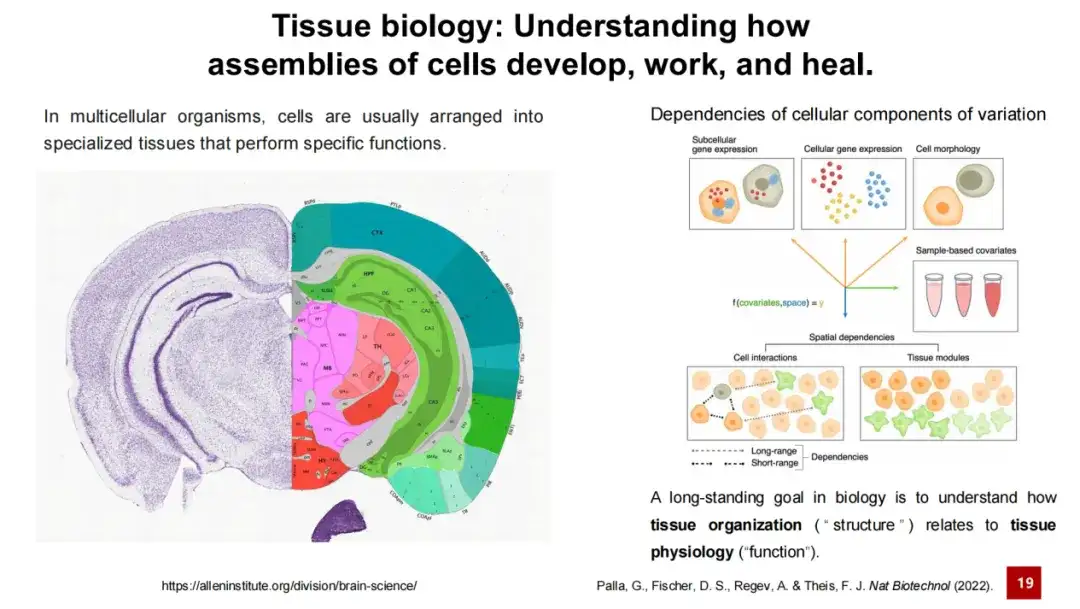
組織研究は常に生命科学研究の中核課題の 1 つであり、組織の構造とその機能の関係を理解することが生物学研究の長期的な目標の 1 つであると言えます。これは容易に理解できます。たとえば、脳内のさまざまな脳領域はさまざまなニューロンと支持細胞で構成され、複雑な細胞間相互作用を通じてさまざまな機能を実行します。たとえば、一部の領域は記憶を担当し、一部の領域は学習を担当し、一部の領域は運動反応を担当します。
したがって、空間トランスクリプトーム解析における中心的な問題は、空間内のさまざまな細胞タイプまたは組織モジュールを識別することです。このタスクは総称して空間クラスタリングと呼ばれます。
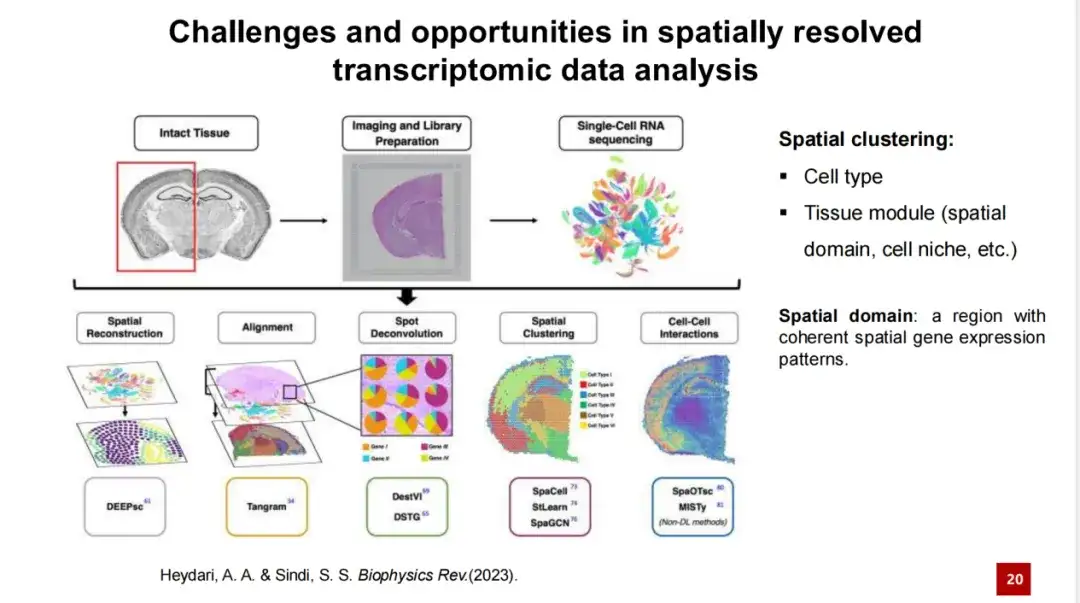
このタスクには 2 つのサブタスクが含まれています。1 つは細胞の種類を識別することであり、もう 1 つは組織モジュール (組織モジュール) を識別することです。。前者はより直観的であり、マウス胚データに示されるような空間トランスクリプトーム データ内のさまざまな細胞タイプを識別します。一方、後者は比較的抽象的で、特定の機能を持つ可能性のある組織構造よりも組織内の小さな領域の識別を伴います。または細胞で構成されています。
さまざまな研究において、研究者は組織モジュール (組織モジュール) に空間ドメイン (空間ドメイン) や細胞微小環境 (細胞ニッチ) などのさまざまな名前を付けますが、その中で空間ドメインの方が一般的に使用される用語です。研究者の中には、組織モジュールを特定することは、一貫した空間的遺伝子発現特性を持つ領域を特定することであると信じている人もいます。
ただし、この概念には限界があります。たとえば、以下の図 A は、2 つの領域間に有意な遺伝子発現の違いがあることを示していますが、図 B および C では、領域間の遺伝子発現分布が完全にきれいではなく、混合している可能性があります。図 B と図 C は、空間領域の概念では解決できない状況を示しています。

これに対して私たちはSPACE法を提案しました。インタラクションを意識したセルの埋め込みを学習することで、空間領域の問題を解決します。
SPACE は、グラフ オートエンコーダー フレームワークを使用して、低次元のセルの埋め込みを学習します。
まず、空間トランスクリプトーム データを入力し、各細胞の空間的位置に基づいて隣接グラフを構築します。つまり、各細胞の最も近い隣接細胞を接続してグラフを形成します。以下の図では、ノードは細胞を表しており、ノードの特性は細胞の遺伝子発現特性です。隣接グラフと遺伝子発現プロファイルを、3 層の GAT ネットワークで構成される SPACE のエンコーダーに入力します。
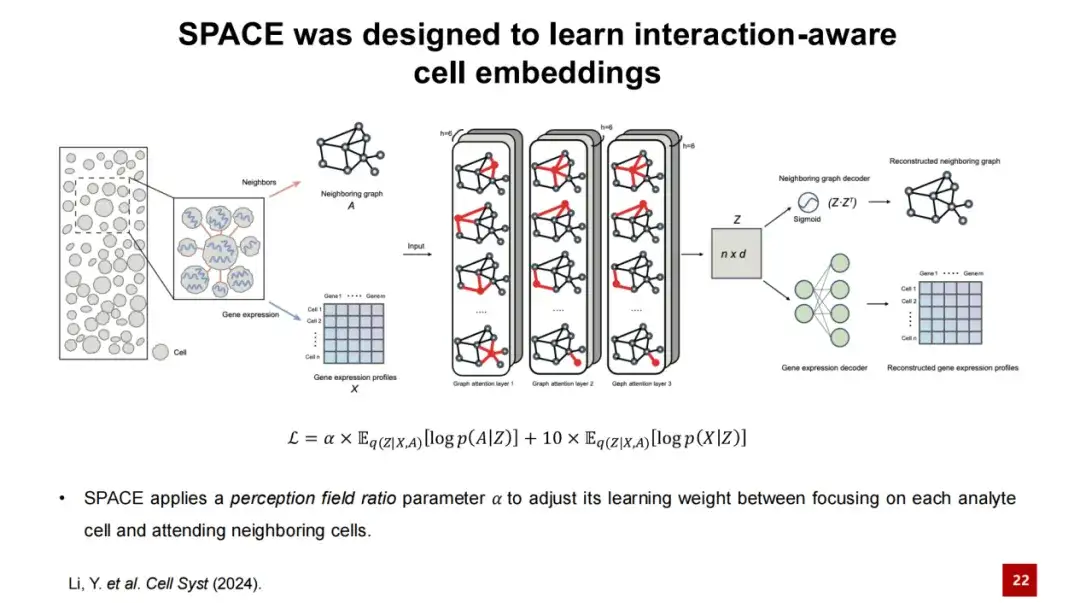
エンコーダーの処理を通じて、各ノードの埋め込み表現を取得し、2 つの独立したデコーダーを通じてそれを再構築できます。1 つのデコーダは細胞の低次元隠れ層表現を近傍グラフに再構築し、もう 1 つのデコーダは細胞の遺伝子発現プロファイルを再構築します。 SPACE モデルの損失関数 (Loss Function) は、これら 2 つの再構成損失の合計です。
このプロセス中に、モデル内の 2 つの損失関数の重みを調整するために、知覚フィールド比パラメーター α (知覚フィールド比パラメーター α) を設計しました。
αの値が小さい場合、このモデルは細胞自体の遺伝子発現の再構築に重点を置いており、この時点で得られた細胞埋め込みは細胞の種類を識別するために使用できます。αの値が大きいと、このモデルは細胞間の相互作用にさらに注目しており、このとき得られる細胞包埋は組織モジュール (Tissue Module) を識別するために使用できます。低次元セル埋め込み Z にはセル相互作用に関する情報が含まれているため、SPACE インタラクションによって得られる低次元埋め込み表現をアワラセル埋め込みと呼びます。
空間細胞のサブタイプを特定するために、マウスの一次運動野データセットに SPACE を適用しました。
下の図では、左上隅に実際の組織内の各細胞の空間的位置が示されており、点が細胞を表し、色が細胞の種類を示しています。これは遺伝子発現に基づいて生成された UMAP マップです。左下の 2 つの図は、SPACE によって識別される空間セルのサブタイプと空間内でのそれらの位置を示しています。これらの空間セル サブタイプと元の研究で提供されたセル タイプ (右図) の混同行列分析を実行したところ、結果は調整されたランド インデックス (ARI) 0.6 と全体的に一致することが示されました。同時に、 SPACE は、星状細胞と希突起膠細胞をより細かく区別し、より多くの細胞サブタイプを識別できます。
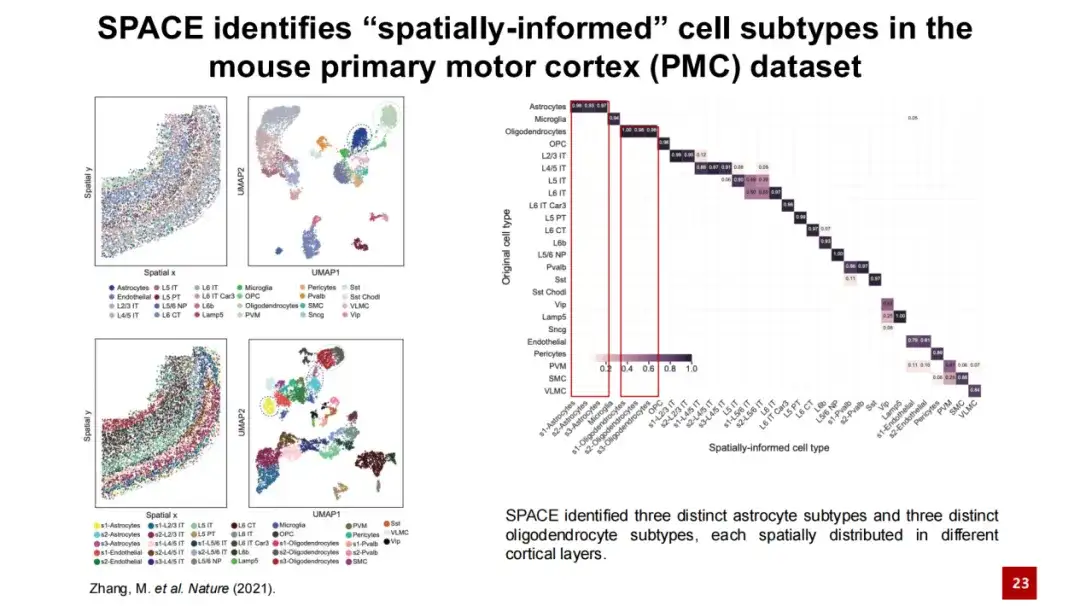
左下の図は、マウスの一次運動野の組織構造を示しています。層は皮質構造を表し、WMは白質を表します。第1層から白質までの層構造がはっきりとわかります。 SPACE によって同定された 3 つの星細胞サブタイプは、遺伝子発現だけでは区別できないため、UMAP プロットでは一緒に混合されています。
ただし、これら 3 つのセル サブタイプは空間分布において明確に区別されています。s1 セル サブタイプは主にレイヤ 1 からレイヤ 4 の領域に分布し、s2 セル サブタイプは主にレイヤ 5 からレイヤ 6 の領域に分布し、s3 セル サブタイプは主にレイヤ 1 からレイヤ 4 の領域に分布しています。ホワイトマター。これら 3 つの星細胞サブタイプの周囲の細胞タイプの割合を数えたところ、結果はこの層別パターンと一致しました。これら 3 つの細胞サブタイプは遺伝子発現において類似していますが、依然として独自の高い発現遺伝子を示します。

SPACE によって同定された 3 つの星細胞サブタイプは、以前の研究と非常に一致しています。これまでの研究では、星細胞とニューロンの間には相互作用があり、星細胞の層状化はニューロンの層状化に対応していることが報告されている。研究者らは、ニューロンの重要な因子をノックアウトすることにより、ニューロンの層状構造が破壊され、それに応じて星細胞の層状構造が変化することを発見した。これは、星細胞とニューロンの間に空間的に特異的な相互作用と空間的に特異的な遺伝子制御があることを示しています。
この例からわかるのは、SPACE は、空間情報を効果的に利用して、空間的特徴を持つさまざまな生物学的細胞の種類を正確に識別できます。
上で紹介したように、SPACE は感知フィールド スケール パラメーター α を調整することでモデルの最適化の方向を変更します。細胞の種類を識別するために細胞自体の特性にもっと注意を払うことも、発見するために細胞間の相互作用情報にもっと注意を払うこともできます。組織モジュール (組織モジュール)。
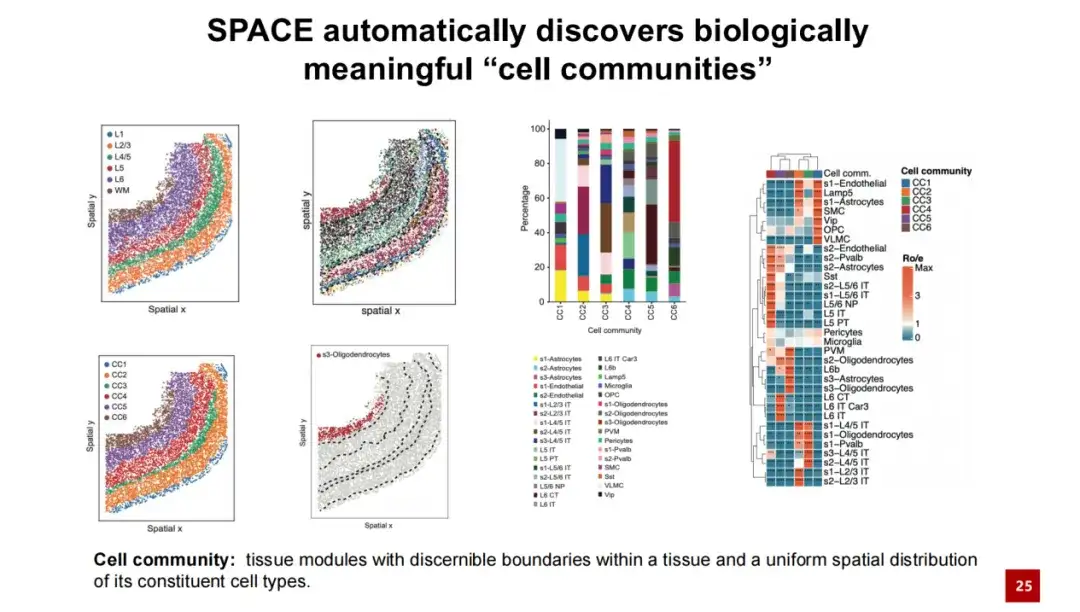
同じデータセット内で、α値を大きくすることで、SPACEは組織モジュールの発見に成功した。私たちはそれをセルコミュニティ(CC)と名付けました。私たちは、SPACE によって発見された組織モジュールには識別可能な境界があり、その中の細胞型の空間分布は比較的均一で一貫していると考えています。 SPACEによって発見された細胞群集と既存の組織構造を比較したところ、両者は1対1でよく対応していることがわかりました。各細胞群には異なる細胞型が含まれており、細胞群内でのこれらの細胞型の空間分布は比較的均一です。
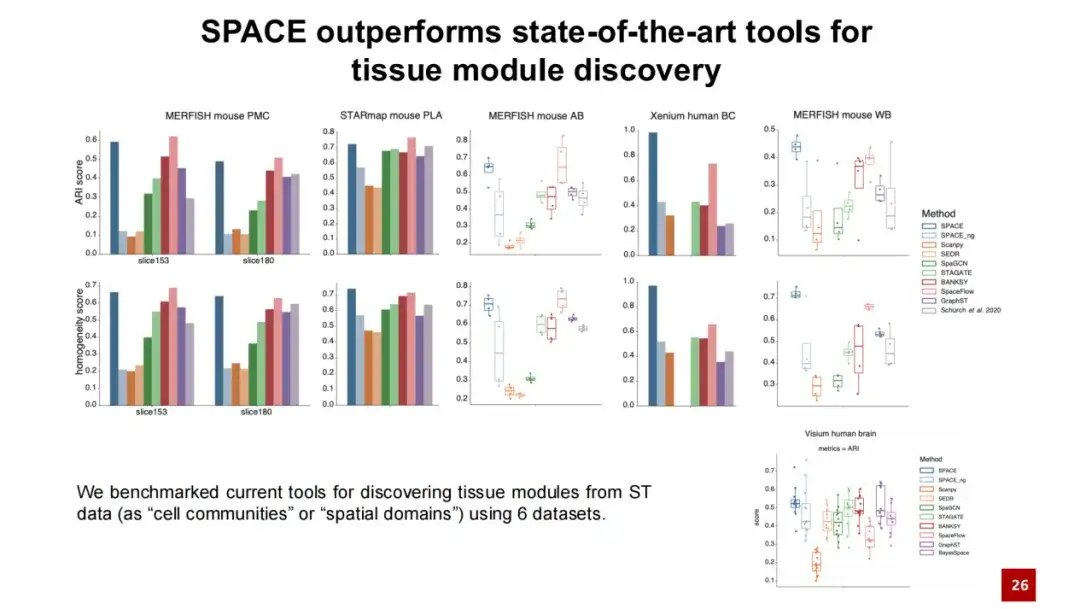
SPACEと組織モジュールを発見できる既存の手法を比較し、5つのデータセットに対してテスト分析を実施しました。結果は、SPACE が 2 つのデータセットでは既存の最良の手法を上回り、他の 3 つのデータセットでは最良の手法と同等であることを示しています。また、一般的に使用されている Visium 人間の脳データセットのテスト分析も実施しました。その結果、SPACE は単一細胞解像度を持たない空間トランスクリプトーム データにも適していることが示されました。
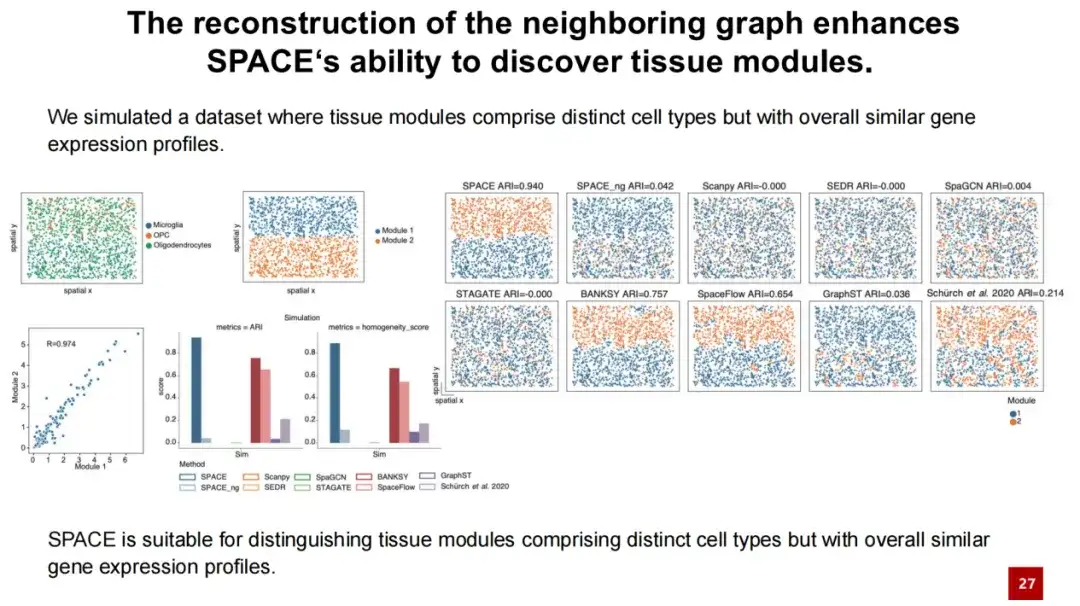
さらに、SPACE_ng という名前のテスト モデルを導入します。ng は、SPACE モデルで隣接グラフ再構成損失をオフにすることを意味します。結果は、SPACE_ng のパフォーマンスが SPACE よりもはるかに低いことを示しています。
SPACE が隣接グラフの再構成から組織モジュールのパフォーマンスをうまく発見できることをさらに説明するために、シミュレーション実験を設計しました。希突起膠細胞と、希突起膠細胞間に均一に分布したミクログリアおよびOPC細胞を選択し(下記左上の画像を参照)、2つの組織モジュールを形成しました。
2 つの組織モジュールの細胞のほとんどは希突起膠細胞であり、類似性が非常に高いため (コラボレーション = 0.97)、SPACE_ng では 2 つの組織モジュールを区別できない一方で、SPACE が他の方法よりもはるかに優れていることがテスト結果からわかります。これは、組織モジュールを識別する際の SPACE のパフォーマンスが、近傍グラフの再構成に由来していることを示しています。
我々は、下流解析でも同様の現象を観察した。つまり、SPACEによって特定された細胞群集の特徴は、空間ドメインのような遺伝子の空間的発現が一貫しているだけではなく、同様の隣接する細胞間の相互作用の違いを反映しているということである。
以下のヒート マップでは、各列がセルを表し、その色はセルが属するセル コミュニティとそのセルの種類を示します。各行はセル タイプを表し、そのセル タイプと他のセルの間の隣接相互作用の相対頻度を示します。このヒート マップから、同じ細胞コミュニティに属する細胞は隣接相互作用において類似性を示し、この類似性は特定の細胞の種類には依存しないことがわかります。対照的に、異なる細胞コミュニティに属する細胞は、隣接相互作用に大きな違いがあります。
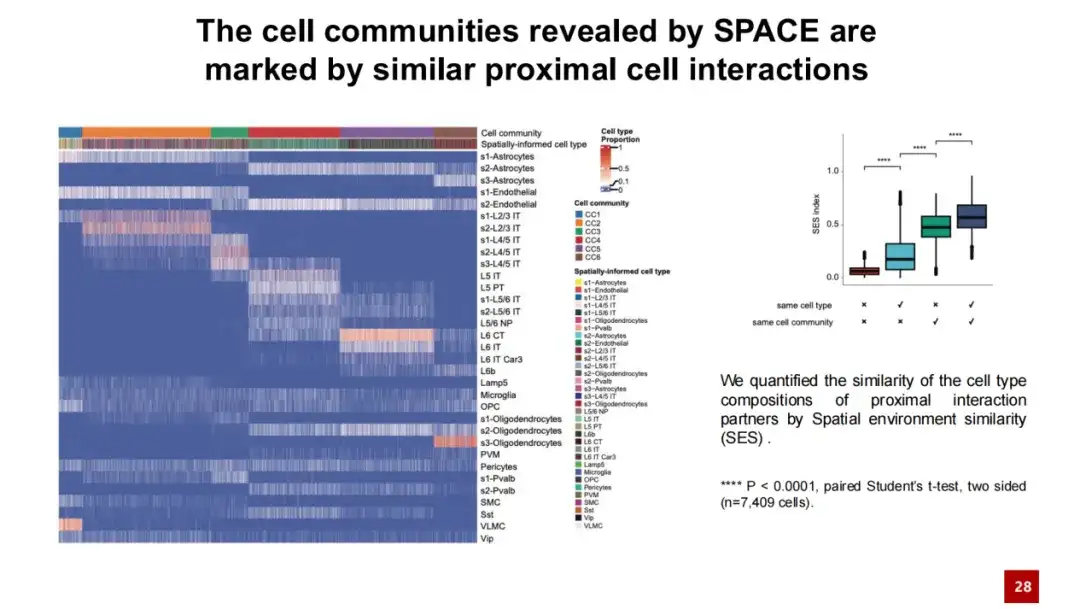
さらにコサイン類似度により細胞間の相互作用の類似度を定量的に計算しました。その結果、同じ細胞群に属する細胞は隣接する細胞間の相互作用に高い類似性がある一方、異なる細胞群に属する細胞は相対的に異なる細胞外相互作用を持つことが示されました。これらの結果は次のことを示していますSPACEによって発見された細胞群集は、遺伝子の空間的発現パターンだけでなく、近位細胞相互作用ネットワークの影響も受けています。
別のマウス胎盤データセットでも同様の分析を実行しました。左の画像はデータセット内の各細胞タイプの空間的位置を示し、中左の画像は手動で注釈が付けられたマウス胎盤組織構造を示し、中右の画像は SPACE によって発見された 5 つの細胞群を示します。 SPACE によって発見された細胞群と手動で注釈が付けられた組織構造の間には、良好な 1 対 1 の対応関係があることがわかります。右側に示すように、各細胞コミュニティごとに特徴的な近位細胞相互作用ネットワークを構築しました。これは、各細胞コミュニティ内での固有の細胞間相互作用を示しています。

CC1 を例に挙げると、このコミュニティは主に母体の脱落膜 (Maternal decidua) 領域に位置しています。我々は、CC1において、s2母体破壊細胞とs2糖栄養膜細胞との間に強い相互作用があることを発見した。これまでの研究では、マウスの妊娠中に糖栄養膜細胞が母体の脱落膜領域に侵入し、その中の母体の復調細胞と相互作用し、それによって母体の血液を胎盤に導く動脈リモデリングプロセスが引き起こされることが示されている。このプロセスは正常な妊娠にとって重要である。
上記の分析から、次のように結論付けることができます。SPACE は、生命プロセスに重要な影響を与える生体サンプル内の細胞間相互作用を特定できます。したがって、私たちは次のように推測しますSPACE によって構築された相互作用ネットワークは、リガンド-受容体ベースの細胞コミュニケーション解析を最適化するために使用できます。
リガンド受容体ベースの細胞コミュニケーション解析は、単一細胞データ解析における一般的な方法であり、2 つの細胞におけるリガンドと受容体の遺伝子発現に基づいて、リガンド受容体ペアを介した細胞コミュニケーションの可能性を推測します。まず、マウス胎盤データセットで CellChat (一般的に使用される細胞通信解析手法) を使用して、CC1 の細胞間通信を解析しました。
CellChatは、s3母体損傷細胞がコラーゲンFN1やTHBSなどのシグナル伝達経路を介してP-TGC細胞型とコミュニケーションできることを発見した。ただし、これらのシグナル伝達経路はすべて、実際に起こるためには物理的な接触を必要とします。しかし、2 つの細胞タイプは実際には空間的に遠く離れており (下図の右下隅を参照)、実際に物理的に接触している可能性は低いことがわかりました。
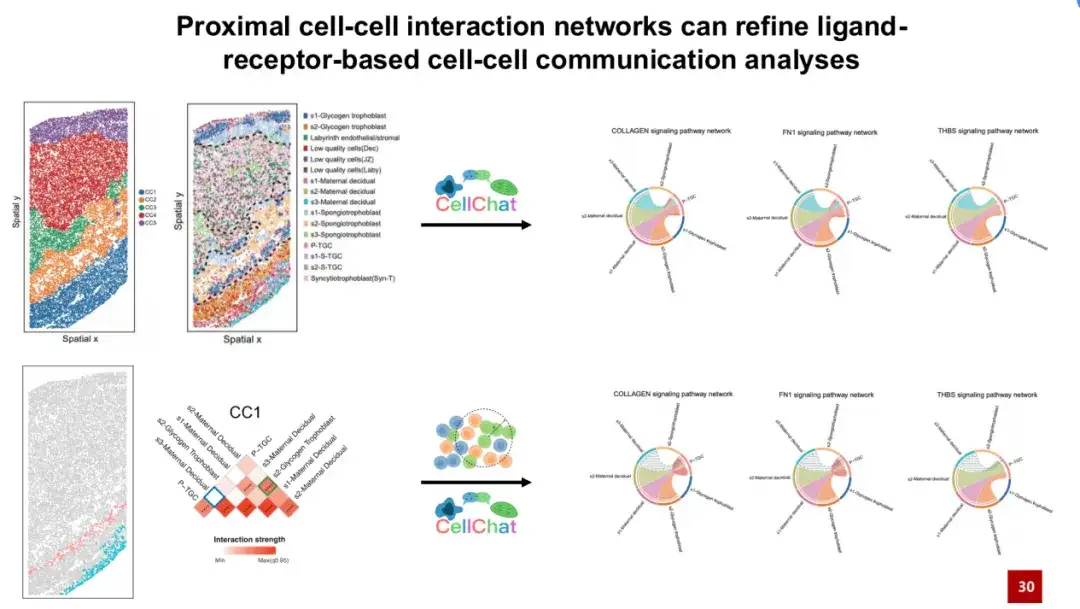
このことは、CC1 で構築された近位細胞相互作用ネットワークによっても確認されました。青いボックスは、それらの間の相互作用が起こりそうもないことを示しています。SPACEによって構築された特徴的な近接細胞相互作用ネットワークをCellChatの細胞通信解析に導入することで、物理的に不可能な細胞通信信号を排除し、誤検知信号を効果的に低減することができます。
募集
清華大学と国家膜生物学重点研究所は杭州に膜構造と人工知能生物学の部門を設立した。現在、チームは人工知能と生物学の間の学際的研究に従事する専門家を募集しています。この分野に興味のある研究者のチームへの参加を心から歓迎します。詳しい募集内容を知りたい方は、下記のQRコードを読み取って詳細をご覧ください。









