Command Palette
Search for a command to run...
ICMLに選ばれました!人民大学チームは等変グラフ ニューラル ネットワークを使用して標的タンパク質の結合部位を予測し、最高のパフォーマンス向上を実現しました 20%
生体システムでは、ほとんどすべての生物学的および薬理学的プロセスに受容体 (標的タンパク質) とリガンド (小分子) 間の相互作用が関与しており、これらの相互作用は標的タンパク質構造の特定の領域で発生します。「結合部位」と呼ばれる - 標的タンパク質の結合部位の予測は、創薬などの下流のさまざまなタスクにおいて基本的な役割を果たします。
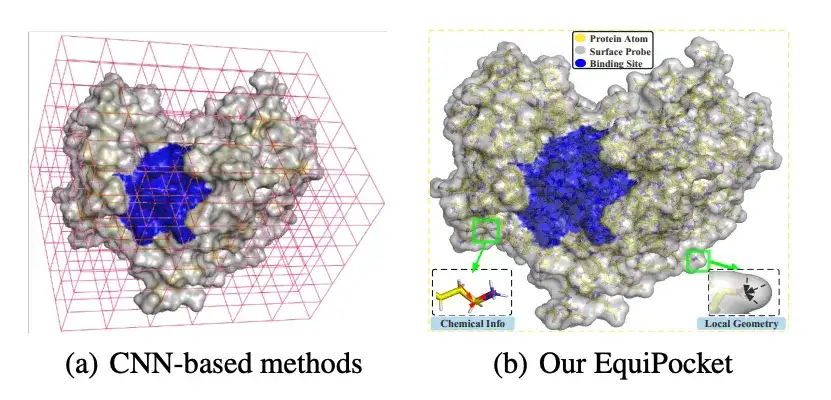
近年、深層学習の画期的な進歩に触発されて、畳み込みニューラル ネットワーク (CNN) がリガンド結合部位の予測に適用されることに成功しました。 CNN ベースの手法は、タンパク質の原子を最も近いボクセルに空間的にクラスタリングすることでタンパク質を 3 次元画像として扱い、結合部位の予測を 3D グリッド上のオブジェクト検出問題またはセマンティック セグメンテーション タスクとしてモデル化します。これらの方法には一定の利点がありますが、まだ課題もあります。不規則なタンパク質構造を表現するのに欠陥があり、回転に敏感であり、タンパク質のサイズの変化に鈍感です。
この目的を達成するために、中国人民大学ヒルハウス人工知能大学院の研究チームは最近、「EquiPocket: リガンド結合部位予測のための E(3) 等変幾何グラフ ニューラル ネットワーク」というタイトルの論文を ICML 2024 で発表しました。 AI分野の学会」の研究論文。この研究では、E(3) 等変グラフ ニューラル ネットワーク (GNN) をリガンド結合部位の予測に初めて適用しました。EquiPocketというフレームワークを提案し、CNN ベースの手法で直面する課題に対処します。
研究のハイライト:
* リガンド結合部位予測に E(3) 等変 GNN を初めて適用
* 従来の CNN ベースの手法と比較して、EquiPocket はボクセル化プロセスを必要とせず、不規則なタンパク質構造をモデル化でき、ユークリッド変換の影響を受けないため、「不規則なタンパク質構造を表現する」という問題を解決します。 「回転に対する感度」
* 代表的なベンチマーク手法に関する広範な実験により、現在の最先端手法に対する EquiPocket の優位性が実証され、創薬などのさまざまな下流タスクを支援します
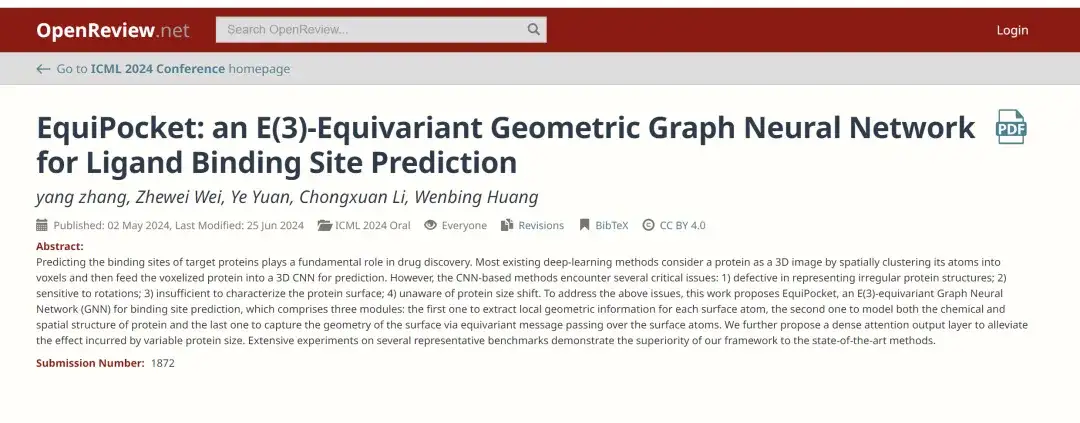
用紙のアドレス:
https://openreview.net/forum?id=1vGN3CSxVs
オープンソース プロジェクト「awesome-ai4s」は、100 を超える AI4S 論文の解釈をまとめ、大規模なデータ セットとツールを提供します。
https://github.com/hyperai/awesome-ai4s
データセット: 複数の専門的なデータセットの包括的な検証
複数の特殊なデータセットを選択し、結合部位の予測に関連するリガンドを含む mlig サブセットを使用して各データセットを評価しました。
で、scPDB は結合部位予測用のよく知られたデータセットです。VolSite によって生成されたタンパク質、リガンド、および 3D キャビティ構造が含まれています。この研究では、トレーニングと相互検証のために 2017 年にリリースされたバージョンを使用しました。このバージョンには、17,594 個の構造、16,034 個のエントリ、4,782 個のタンパク質、および 6,326 個のリガンドが含まれています。
PDBbind は、タンパク質-リガンド複合体を研究するために一般的に使用されるデータセットです。タンパク質、リガンド、結合部位の 3D 構造、および実験室で測定された正確な結合親和性の結果が含まれています。この研究は、通常セット (14,127 複合体) と洗練されたセット (5,316 複合体) の 2 つの部分で構成される 2020 年バージョンを使用して評価されました。共通セットにはすべてのタンパク質-リガンド複合体が含まれており、洗練されたセットでは、実験テスト用に共通セットからより品質の高い化合物が選択されます。
COACH 420 と HOLO4K は、結合部位の予測に使用される 2 つのテスト データ セットです。最初に導入されたのは (Krivák & Hoksza、2018) です。
モデル アーキテクチャ: EquiPocket の全体的なフレームワークは 3 つの主要なモジュールで構成されています
EquiPocket の全体的なフレームワークは 3 つのモジュールで構成されます。以下に示すように:
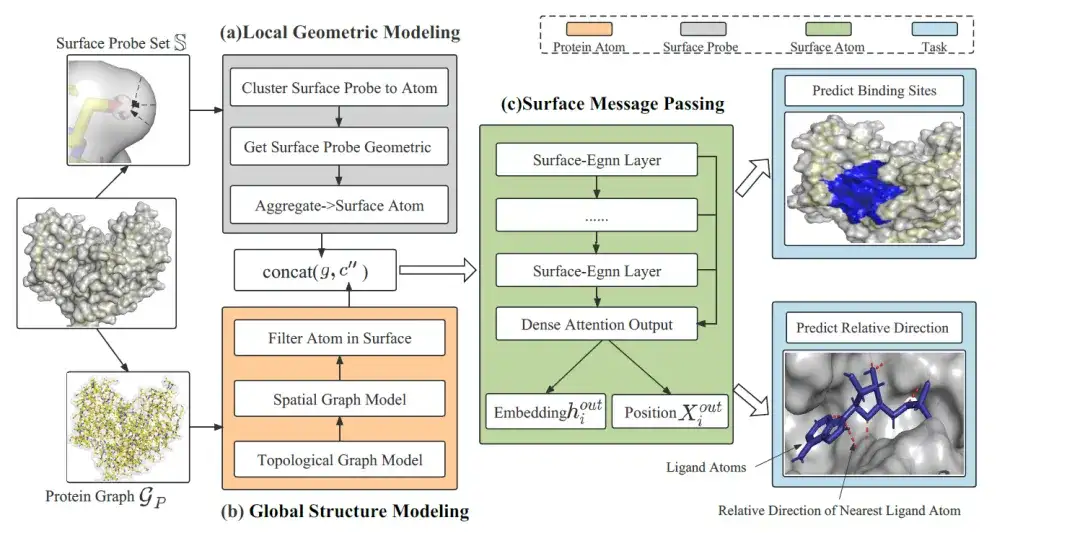
最初のモジュールはローカル幾何モデリング モジュール (ローカル幾何モデリング) で、各表面原子のローカル幾何情報を抽出するために使用されます。2 番目のモジュールはグローバル構造モデリング モジュール (グローバル構造モデリング) で、タンパク質の化学と空間構造、最後のモジュールは表面メッセージ パッシング モジュール (表面メッセージ パッシング) で、表面原子の等変情報を送信することで表面の形状を捕捉します。
ローカルジオメトリモデリングモジュール
各タンパク質原子の局所的な幾何学的形状によって、その近くの領域が結合部位の一部となるのに適しているかどうかが決まります。
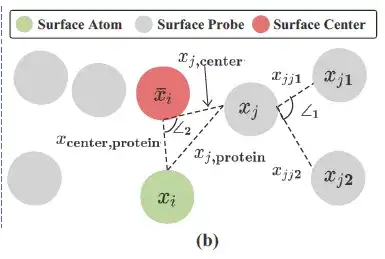
上の図に示すように、研究者は各タンパク質表面原子 (上図の緑色) の周囲に表面プローブ (表面プローブ (上図の灰色)) を使用して、局所的な幾何学的情報を記述します。具体的には、各表面原子 i ∈ VS について、その周囲の表面プローブが S のサブセットによって返されます。つまり、次のようになります。
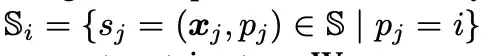
研究者らは、Si に基づいて幾何学的情報を構築し、Si 内のすべての 3D 座標の中心/平均値 (Surface Cente、上の図の赤色) を xi として記録します。
グローバル構造モデリングモジュール
結合部位は主に表面原子で構成されていますが、タンパク質の全体構造はリガンド相互作用や結合部位の形成に影響を与えることが多いため、モデル化する必要があります。
研究者らは、化学グラフ モデリングと空間グラフ モデリングという 2 つの接続されたプロセスを通じてこの目標を達成しました。結果として得られるグローバル構造モデリング モジュールは、原子の種類、化学結合、関連する空間位置などを含むタンパク質全体の情報の処理を担当します。
表面情報伝達モジュール
表面原子の局所的な幾何学的特徴とグローバルなエンコード特徴を考慮すると、このモジュールは表面マップ上で等変情報転送を実行して、タンパク質表面原子のすべての特徴を更新します。
研究結果: EquiPocket はベースライン モデルと比較して 10-20% のパフォーマンス向上
実験では、研究者らは EquiPocket と比較するために次のベースライン モデルを使用することを選択しました。
* ジオメトリベースの手法: Fpocket
※機械学習手法:P2rank
* CNN ベースのメソッド: DeepSite、Kalasanty、DeepSurf、RecurPocket
* トポロジー グラフベースのモデル: GAT、GCN、および GCN2
* 空間グラフベースのモデル: SchNet、EGNN
モデルの評価に使用されるメトリクスには、DCC (予測された結合部位中心と真の結合部位中心との間の距離)、DCA (予測された結合部位中心と任意のリガンド グリッドとの間の最短距離)、および失敗率 (予測された結合部位中心がない場合のサンプリング率) が含まれます。以下の表はCOACH 420、HOLO4K、PDBbindの結合部位予測結果です。
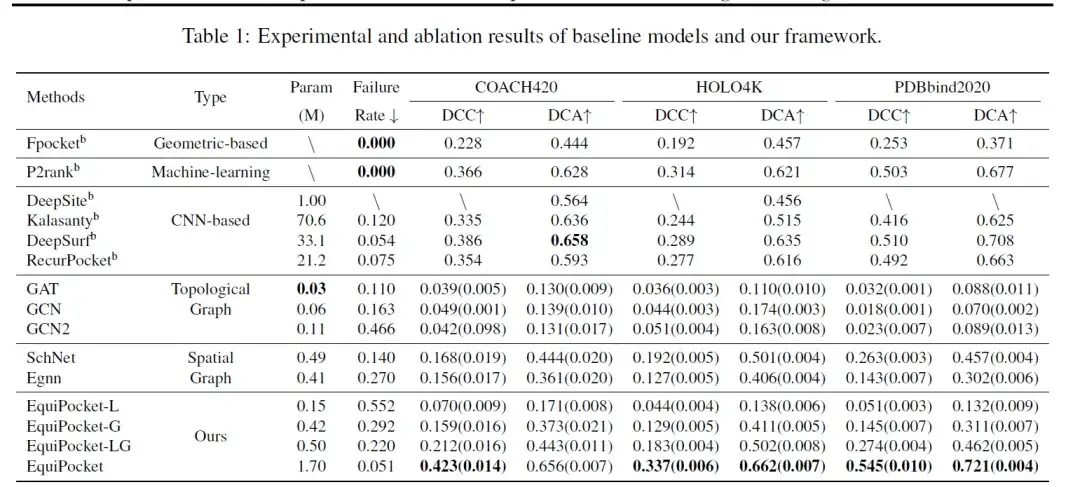
表のデータに示されているように、ジオメトリベースのメソッド Fpocket のパフォーマンスは低く、この方法はタンパク質の幾何学的特徴のみを使用するため、失敗率は 0 です。機械学習方法 P2rank は、ランダム フォレストとタンパク質表面の幾何学的情報を組み合わせることで、パフォーマンスを大幅に向上させます。
CNN ベースのメソッド (DeepSite、Kalasanty、DeepSurf、RecurPocket) のパフォーマンスは、ジオメトリ ベースのメソッドのパフォーマンスよりもはるかに高くなります。このうち DCC と DCA は 50% よりも改良されていますが、多くのパラメータと計算リソースを必要とします。このうち、初期に提案された手法である DeepSite と Kalasanty は、タンパク質のサイズ変化や大きなタンパク質を扱う能力が不十分であるという制限があり、予測の失敗につながる可能性があります。
グラフィカルモデルの場合、トポロジカル グラフ モデル (GCN、GAT、GCN2) のパフォーマンスが低く、主な理由は、原子および化学結合の情報のみを考慮し、タンパク質の空間構造を無視するためです。通常、空間グラフ モデル (SchNet、EGNN) のパフォーマンスはトポロジカル グラフ モデルよりも優れています。 EGNN は原子のプロパティとその相対/絶対空間位置を使用するため、より適切に機能します。SchNet は原子の相対距離に基づいて埋め込みを更新するだけですが、空間グラフ モデルのパフォーマンスは CNN ベースおよびジオメトリ ベースの方法よりも劣ります。前者は十分な幾何学的特徴を得ることができず、タンパク質のサイズ変化の問題も解決できません。
上記の結果は次のことを示しています。結合部位の予測には、タンパク質表面の幾何学的情報と多層構造情報が重要です。
さらに、これは現在の GNN モデルの限界を反映しています。つまり、タンパク質表面から十分な幾何学的情報を収集することが難しい、または必要な計算リソースが大きすぎてタンパク質などの高分子システムに適用できないことです。したがって、EquiPocket フレームワークは、原子レベルから化学的および空間的情報を更新するだけでなく、過剰なコンピューティング リソースを必要とせずに幾何学的情報を効率的に収集します。そのパフォーマンスは、以前の結果よりも 10-20% を向上させています。
低分子リガンドから生体高分子まで、AI がタンパク質構造を深く解釈
すべての植物、動物、人間の細胞の内部には、タンパク質、核酸、糖、その他の分子で構成される何十億もの分子機械があり、それらのどの部分も単独で機能することはできません。相互作用を通じてのみ、それらが何百万もの異なる中でどのように機能するかを理解する必要があります。組み合わせることで、人生をより深く理解できるようになります。
今年 5 月、Google DeepMind は、タンパク質、核酸、小分子、イオン、修飾残基を含む複合体の結合構造を予測できる AlphaFold3 モデルをリリースしました。タンパク質と小分子リガンド間の相互作用は、薬物の作用メカニズムの中核です。AlphaFold3 は、高度な深層学習アルゴリズムを通じて、既存のドッキング ツールをはるかに上回る精度で、タンパク質とリガンドの結合の三次元構造を正確に予測できます。 。
新薬開発に関しては、AlphaFold3 によって予測されるタンパク質 - リガンド構造を通じて、研究者は新薬候補をより効果的にスクリーニングおよび設計し、既存薬の最適化という観点から創薬プロセスを加速することができます。このツールは、既存薬との相互作用を改善することによって、既存薬を最適化するためにも使用できます。有効性を高めたり、副作用を軽減したりするためのタンパク質結合パターン。
低分子リガンドに加えて、タンパク質の生物学的機能は、DNA や糖などの生物学的高分子と組み合わせる必要もあります。現在、実験的方法により何千ものタンパク質構造複合体がタンパク質データベースに登録されていますが、従来の実験的方法は時間と費用がかかり、機械学習に基づく予測方法はこの課題を簡単に解決できます。
今年 2 月、南京農業大学の研究チームは、「ULDNA: 高精度のタンパク質 DNA 結合部位予測のための教師なしマルチソース言語モデルと LSTM アテンション ネットワークの統合」というタイトルの記事を、重要なバイオインフォマティクスのブリーフィングにオンラインで発表しました。生物学分野のジャーナル」の研究論文。タンパク質とDNAの結合部位予測の問題を目指して、新しいディープラーニング予測手法ULDNAが開発されました。
用紙のアドレス:
https://academic.oup.com/bib/article/25/2/bbae040/7606634

ULDNA の核となるアイデアは、タンパク質大言語モデルを使用して配列の特徴表現を設計し、その後、アテンション メカニズムの長期短期記憶ネットワーク (LSTM-Attention Network) を組み合わせて DNA 結合部位予測モデルをトレーニングすることです。 。研究者らは、ULDNA に関する包括的なテストを行うために、PDNA-128、PDNA-316、および PDNA-335 を含む 7 つのベンチマーク データ セット (タンパク質配列の数は 40 ~ 600 の範囲) を選択しました。実験結果は次のことを示していますULDNA はすべてのデータセットで優れたパフォーマンスを発揮し、その予測パフォーマンスは他の 9 つの主流の方法よりも大幅に優れています。
DNA を超えて、糖はすべての生物の細胞表面に遍在し、そこでレクチン、抗体、酵素、トランスポーターなどの複数のタンパク質ファミリーと相互作用して、免疫応答、細胞分化、神経発達などの重要な生物学的プロセスを調節します。炭水化物とタンパク質の間の相互作用メカニズムを理解することは、炭水化物医薬品を開発するための基礎です。しかし、炭水化物の構造の多様性と複雑さ、特にタンパク質との結合部位の多様性は、実験データの取得と医薬品設計に課題をもたらしています。
つい最近、中国科学院のチームは、特定のタンパク質構造上の糖結合部位を正確に予測できる深層学習モデルである DeepGlycanSite を開発しました。 DeepGlycanSite は、タンパク質の幾何学的および進化的特性を、Transformer アーキテクチャに基づいた深い等変グラフ ニューラル ネットワークに統合します。その性能はこれまでの先進的な手法を大幅に上回り、さまざまな糖分子の結合部位を効果的に予測できます。
DeepGlycanSite は、突然変異誘発研究と組み合わせることで、重要な G タンパク質共役受容体のグアノシン-5'-二リン酸認識部位を明らかにします。これらの発見は、糖結合部位の予測における DeepGlycanSite の価値を実証し、治療上重要なタンパク質の糖調節の背後にある分子機構への洞察を提供する可能性があります。

この研究は「DeepGlycanSiteによる高精度炭水化物結合部位予測」と題され、2024年6月17日にNature Communications誌に掲載された。
用紙のアドレス:
https://www.nature.com/articles/s41467-024-49516-2
タンパク質は生物にとって重要な分子であり、細胞の構造と機能において重要な役割を果たしています。タンパク質の構造を研究することは、生命過程の理解、病気のメカニズムの解明、薬の開発にとって非常に重要です。今日、機械学習は科学者に生命の謎を理解するための新たな扉を開きました。
参考文献:
1.https://openreview.net/forum?id=1vGN3CSxVs
2.https://mp.weixin.qq.com/s/aGzcr0ncQA-jBy-vTGC35Q
3.https://www.jiqizhixin.com/articles/2024-05-09
4.https://news.njau.edu.cn/2024/0








