Command Palette
Search for a command to run...
ニューラル ネットワークが密度汎関数理論に置き換わります。清華大学の研究グループが超高精度の予測を実現するユニバーサル材料モデル DeepH をリリース

材料設計では、その電子構造と特性を理解することが、材料性能の予測、新材料の発見、材料性能の最適化の鍵となります。過去、密度汎関数理論 (DFT) は、材料の電子構造と特性を研究するために業界で広く使用されています。その本質は、分子 (原子) の基底状態におけるすべての情報の伝達物質として電子密度を使用することです。単一電子の波動関数の代わりに、複数電子系を単一電子問題に変換して解くことで、計算プロセスが簡素化されるだけでなく、計算精度が確保され、細孔径分布をより正確に反映することができます。
ただし、DFT は計算コストが非常に高く、多くの場合、小規模な材料システムの研究にしか使用できません。 Materials Genome Initiative に触発されて、科学者たちは DFT を使用して巨大な材料データベースを構築しようと試み始めましたが、これまでに収集されたデータセットは限られていますが、これはすでに素晴らしいスタートとなっています。これを皮切りに、AI技術がもたらす新たな変化を受けて、研究者たちは「ニューラルネットワークにDFTの本質を深く学習させるディープラーニングとDFTを組み合わせることで、革命的なブレークスルーをもたらすことができるのではないか」と考え始めた。
これは、深層学習密度汎関数理論ハミルトニアン (DeepH) メソッドの中核です。DFT の複雑さをニューラル ネットワークにカプセル化することで、DeepH は前例のない速度と効率で計算できるだけでなく、トレーニング データが増加するにつれてインテリジェンスも向上し続けます。最近、清華大学物理学科の Xu Yong 氏と Duan Wenhui 氏の研究チームは、独自の DeepH 手法を使用して DeepH ユニバーサル材料モデルを開発することに成功し、「大規模な材料モデル」を構築するための実現可能なソリューションを実証しました。この画期的な進歩は次のとおりです。革新的な材料の発見は新たな機会をもたらします。
関連する研究は「深層学習密度汎関数理論ハミルトニアンの普遍的材料モデル」というタイトルで、Science Bulletinに掲載されました。

用紙のアドレス:
https://doi.org/10.1016/j.scib.2024.06.011
オープンソース プロジェクト「awesome-ai4s」は、100 を超える AI4S 論文の解釈をまとめ、大規模なデータ セットとツールを提供します。
https://github.com/hyperai/awesome-ai4s
AiiDAを通じて大規模な材料データベースを構築し、磁性材料からの干渉をターゲットを絞って排除します。
DeepH ユニバーサル材料モデルの普遍性を証明するために、この研究では、自動インタラクティブ インフラストラクチャおよびデータベース (AiiDA) を通じて 104 の固体材料を含む大規模な材料データベースを構築しました。
多様な元素組成を実証するために、研究では元素の周期表の最初の 4 行も選択し、これにより磁性材料からの干渉を避けるために Sc から Ni への遷移元素を除外し、希ガス元素も除外しました。候補材料構造は Materials Project データベースから派生します。元素タイプに基づくフィルタリングに加えて、材料プロジェクトでは候補材料がさらに絞り込まれ、「非磁性」とラベル付けされたもののみが含まれます。簡単にするために、単位格子に 150 個を超える原子を含む構造は除外されます。
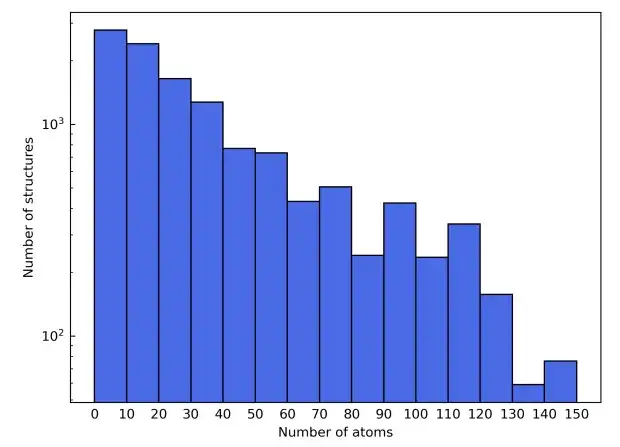
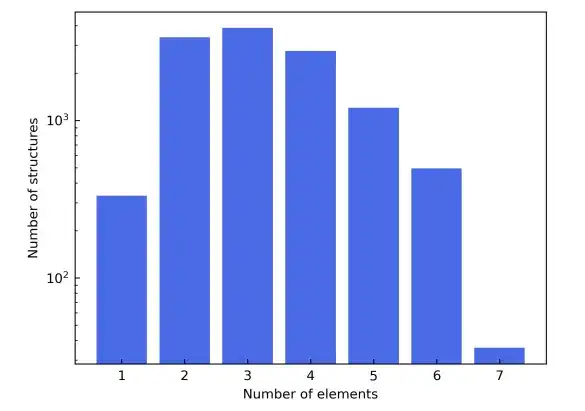
これらのフィルタリング基準の結果、最終的なマテリアル データセットは合計 12,062 の構造で構成されます。トレーニング プロセス中、データ セットは 6:2:2 の比率でトレーニング セット、検証セット、テスト セットに分割されます。次、この研究では、AiiDA (Automated Interactive Infrastructure and Database) のフレームワークを利用して、密度汎関数理論の計算を実行し、それを材料データベースの構築に使用するための高スループットのワークフローを開発します。
DFT ハミルトニアンをターゲットとして、DeepH-2 メソッドを使用して DeepH をトレーニングします
研究では次のように考えられていますDFT ハミルトニアンは理想的な機械学習のターゲットです。
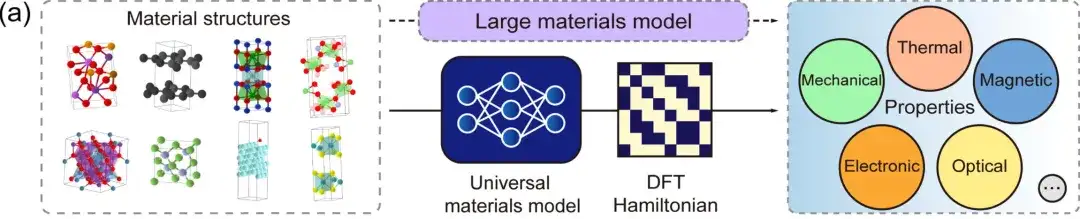
初め、DFT ハミルトニアンは、総エネルギー、電荷密度、バンド構造、物理的応答などの物理量から直接導出できる基本量です。DeepH ユニバーサル材料モデルは、任意の材料構造を入力として受け入れ、対応する DFT ハミルトニアンを生成できるため、上図に示すように、さまざまな材料特性を直接導き出すことができます。
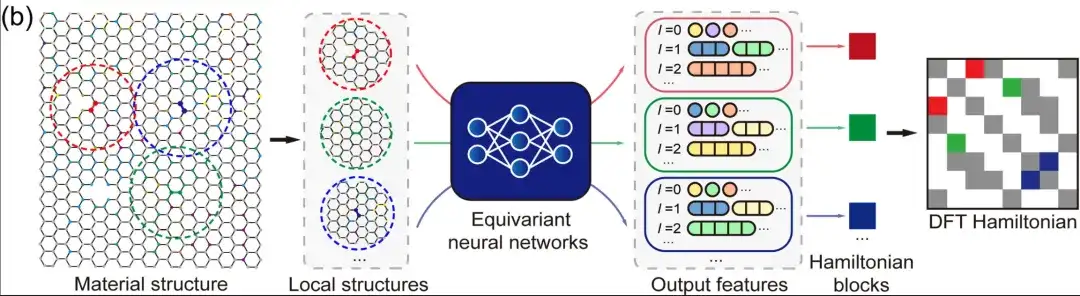
第二に、局所的な原子基底セットの下では、DFT ハミルトニアンは、行列要素が局所的な化学環境によって決定される疎行列として表現できます。等変ニューラル ネットワークでは、上の図に示すように、DeepH はさまざまな角量子数 l でマークされた出力特徴を使用して DFT ハミルトニアンを表します。したがって、材料構造全体の DFT ハミルトニアン行列をモデル化する必要がなく、近くの構造情報に基づいて原子のペア間のハミルトニアン行列要素をモデル化できます。これにより、深層学習タスクが大幅に簡素化されるだけでなく、トレーニング データの量も大幅に増加します。推論側では、深層学習ネットワークが十分なトレーニング データを学習すると、トレーニングされたモデルは、より新しい未知の材料構造にうまく一般化できます。
DeepH の重要なアイデアは、ニューラル ネットワークを使用して HDFT を表現することです。入力マテリアルの構造を変更することにより、DFT コードによって生成された HDFT トレーニング データが最初に作成され、次にニューラル ネットワークのトレーニングに使用されます。これらの訓練されたネットワーク モデルは、新しい材料構造を推論するために使用されます。
このプロセスでは、2 つの非常に重要なアプリオリな知識があります。1 つは局所性の原理、この研究は、原子の局所サンプルにおける DFT ハミルトニアンを表し、ハミルトニアンを原子間結合または原子内結合を記述するブロックに分解します。したがって、単一のトレーニング教材構造は、大量のデータのハミルトン ブロックに対応する可能性があります。さらに、各ハミルトニアン ブロックは、構造全体ではなく局所構造に関する情報に基づいて決定できます。この簡素化により、DeepH モデルの高精度と転送可能性が保証されます。
2つ目は対称性の原理です。物理法則は、異なる座標系から見ても同じままです。したがって、対応する物理量と方程式は、座標変換の下で等価性を示します。等価性を維持すると、データ効率が向上するだけでなく、汎化能力も強化され、DeepH のパフォーマンスが大幅に向上します。第一世代 DeepH アーキテクチャは、ローカル座標系を通じて等価性問題を単純化し、ローカル座標の変換を通じて等価特徴を復元します。第 2 世代 DeepH アーキテクチャは同等のニューラル ネットワークに基づいており、DeepH-E3 と呼ばれます。このフレームワークでは、すべての入力層、隠れ層、出力層の特徴ベクトルは同等のベクトルです。最近、この研究の著者の 1 人が、深層学習のための新世代アーキテクチャである DeepH-2 を提案しました。効率と精度の点では、DeepH-2 が最高のパフォーマンスを発揮します。
要約すると、この研究の深層学習モデル DeepH は DeepH-2 メソッドを使用してトレーニングされ、合計 1,728 万個のパラメーターが含まれており、それぞれのメッセージ パッシングに使用できるニューラル ネットワークを形成します。ノードとエッジは 80 個の等しい価格特性を持ちます。材料構造の埋め込みには、ガウス平滑法を使用した原子番号と原子間距離が含まれ、基底関数の中心範囲は 0.0 ~ 9.0 Å です。ニューラル ネットワークの出力特徴は線形層を通過し、次に DFT ハミルトニアンが Wigner-Eckart 層を通じて構築されます。
この調査は、NVIDIA A100 GPU で合計 343 エポックにわたってトレーニングされ、207 時間かかりました。トレーニング プロセス全体を通じて、バッチ サイズは 1 に固定されます。これは、各バッチに 1 つの材料構造が含まれることを意味します。最後に、初期学習率が 4×10-4、減衰率が 0.5、減衰忍耐力が 20 の場合、選択された最小学習率は 1×10-5 であり、学習率がこの値に達するとトレーニングは停止されます。 。
DeepH は優れた推論パフォーマンスを備え、正確なバンド構造予測を提供します。
トレーニング、検証、およびテスト セットでは、モデルによって予測された密度汎関数理論のハミルトニアン行列要素の平均絶対誤差 (MAE) は、それぞれ 1.45、2.35、および 2.20 meV に達しました。これは、目に見えない構造について推論するモデルの能力を示しています。
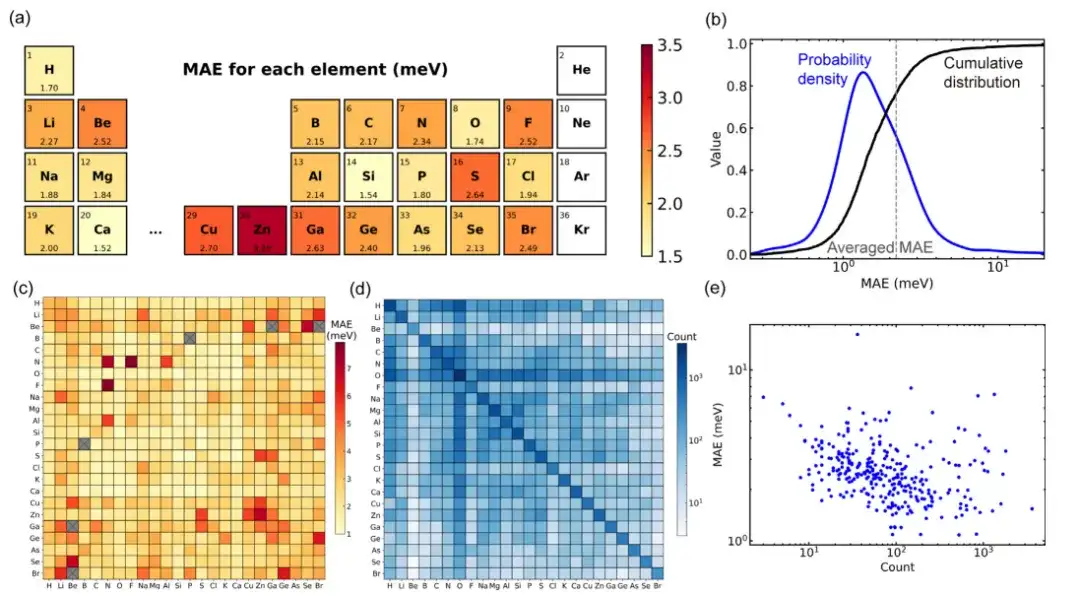
104 個の固体材料からなる大規模な材料データベースを使用して Deep-2 メソッドでトレーニングされたユニバーサル材料モデルのパフォーマンスを評価すると、データセット内のすべての構造のうち、材料構造の約 80% の平均絶対値が平均値 ( 2.2meV)エラー。わずか 34 個の構造 (テスト セットの約 1.4%) の平均絶対誤差が 10meV を超えており、このモデルが主流の構造に対して良好な予測精度を備えていることを示しています。
データセットをさらに分析すると、材料構造に対するモデルのパフォーマンスの偏差が、データセットの分布の偏差に起因している可能性があります。この研究では、データセットに含まれる要素ペアのトレーニング構造が多いほど、対応する平均絶対誤差が小さくなることがわかりました。この現象は、深層学習の一般的な材料モデルに「スケーリング則」が存在することを示している可能性があります。つまり、トレーニング データ セットが大きいほどモデルのパフォーマンスが向上する可能性があります。
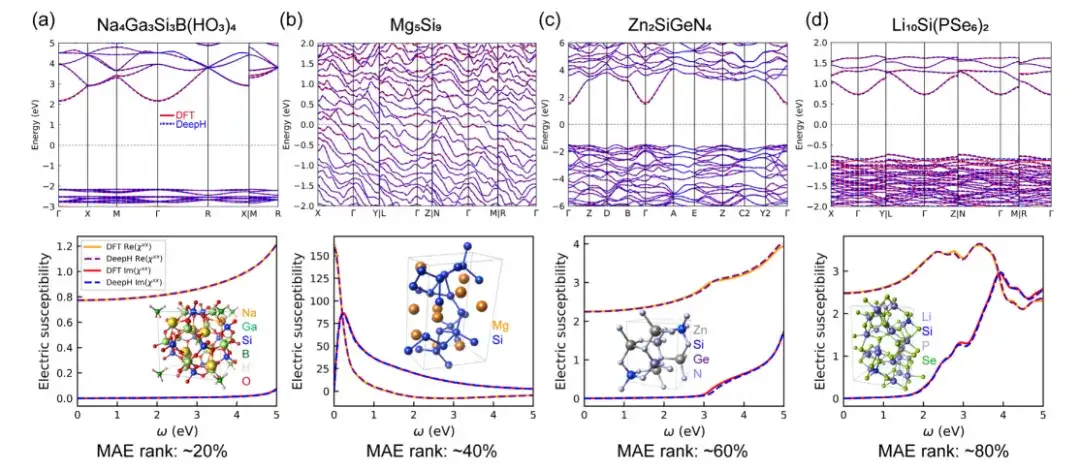
材料特性を予測する際の DeepH ユニバーサル材料モデルの精度を評価するために、この研究では、例の計算時に密度汎関数理論 (DFT) 計算と DeepH 予測 DFT ハミルトニアンを使用し、これら 2 つの方法で得られた計算を組み合わせました。を比較した。結果は次のことを示していますDeepH によって予測された結果は、DFT によって計算された結果に非常に近く、材料特性の計算における DeepH の優れた予測精度を示しています。
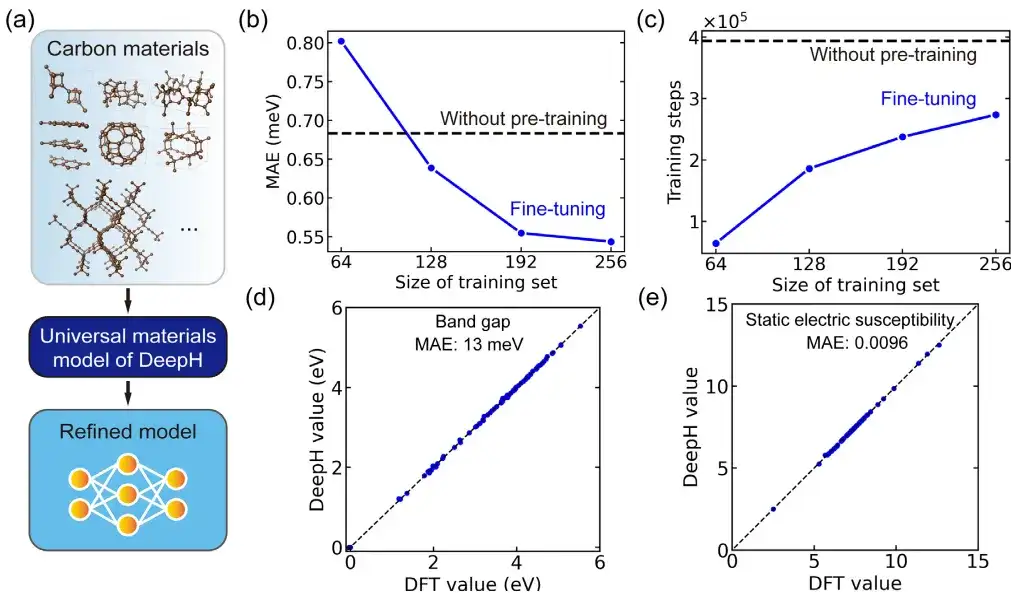
この研究では、特定の用途において、微調整された普遍的な材料モデルを使用して炭素の同素体を調査しました。その中で、炭素材料データセットはサマラ炭素同素体データベース (SACADA) から取得されており、異なる原子構造を持つ合計 427 個の炭素同素体が含まれています。
これに基づいて、研究者らは一般的な材料モデルを微調整し、カーボン材料専用に改良された DeepH モデルを作成しました。事前トレーニングなしのモデルと比較して、微調整により DFT ハミルトニアンの予測における平均絶対誤差を 0.54 meV まで大幅に削減でき、50% 未満のトレーニング構造でも同等の予測精度を達成できます。
さらに、微調整によりトレーニングの収束が大幅に向上し、トレーニング時間が短縮されます。微調整することで予測精度の向上やトレーニング効率の向上につながると言えます。さらに、微調整された DeepH モデルは、材料特性の予測において大きな利点を示し、テストされたほぼすべての構造に対して正確なバンド構造を予測できます。
大規模な材料モデルが急増しているが、AI4S の実現には長い道のりがある
ChatGPT を出発点として、AI は正式に新たな「大型モデル時代」に突入しました。この時代の特徴は、複雑なタスクを処理できる深層学習モデルをトレーニングするために、巨大なデータセットと高度なアルゴリズムを使用することです。材料科学の分野では、これらの大型モデルと研究者の英知が融合し、これまでにない新たな研究の時代が開かれています。これらの大規模モデルは、膨大な科学データを処理および分析できるだけでなく、材料の特性や挙動を予測することもできるため、新材料の発見と開発が加速され、この分野の効率化と正確化が促進されます。
過去の期間において、科学用 AI は常に物質科学と衝突して新たな火花を生み出してきました。
国内をベースに、北京国立物性物理学研究センターの SF10 グループ、中国科学院物理研究所、および中国科学院コンピュータ ネットワーク情報センターは協力して、数万の化学合成経路データを大型モデルLLAMA2-7bにより、材料の無機合成経路の予測に使用できるMatChatモデルの取得に成功、中国電子科学技術大学、復丹大学、中国科学院寧波材料技術工学院が成功世界に先駆けて「耐疲労性強誘電体材料」を開発し、70の課題を解決しました。強誘電体材料の疲労は長年の問題であり、上海交通大学の AIMS 研究室は、新世代の材料インテリジェント設計モデル Alpha Mat を開発しました。研究結果は頻繁に発表されており、材料の革新と発見は新たな時代に入りました。
世界的に見ても、Google の子会社である DeepMind は、材料科学用の人工知能強化学習モデルである GNoME を開発し、熱力学的に安定した結晶材料を 380,000 個以上発見しました。これは、「人類に 800 年間の知的蓄積を加えたもの」に相当し、物質の発見を大幅に加速させます。新しい材料研究のスピード; Microsoft が材料科学分野でリリースした人工知能生成モデルである MatterGen は、必要な材料特性に基づいて新しい材料の構造を予測できます。業界トップの触媒材料データセット Open Catalystプロジェクト、および有機金属フレームワーク吸着データセット OpenDAC... テクノロジーの巨人は、独自のテクノロジーを使用して材料科学の分野をかき立ててきました。
従来の材料の研究開発手法と比較すると、人工知能はより広範囲の材料の可能性を探求する扉を開き、材料の発見に関連する時間と費用を大幅に削減します。しかし、科学のための AI は、材料分野での信頼性と効果的な実装という課題にも直面しており、データ品質の確保、AI システムのトレーニングに使用されるデータの潜在的なバイアスの特定と軽減などの一連の問題を解決する必要があります。これはまた、人工知能が材料科学の分野でより大きな役割を果たすことができるようになるまでには、まだ長い道のりがあることを意味しているのかもしれません。








