Command Palette
Search for a command to run...
ICML、清華航空などのトップカンファレンスに選出され、従来のSOTAを超えるタンパク質言語モデルESM-AAを共同リリース

細胞内の無数の生化学反応の原動力として、タンパク質は細胞の微細な世界で建築家やエンジニアの役割を果たし、生命活動を触媒するだけでなく、生物の形態と機能を構築し維持するための基本的な構成要素でもあります。 。生命の壮大な青写真を支えるのは、タンパク質間の相互作用と相乗効果です。
しかし、タンパク質の構造は複雑で変化しやすく、従来の実験方法ではタンパク質の構造を解析するには時間と労力がかかります。この歴史的な瞬間に、大量のデータを分析することでタンパク質を学習するディープラーニング技術を使用するタンパク質言語モデル (PLM) が登場しました。タンパク質配列データと共進化モデルは、タンパク質の構造予測、適応性予測、タンパク質設計の分野で目覚ましい成果を上げ、タンパク質工学の発展を大きく促進しました。
PLM は残留物スケールでは大きな成功を収めていますが、原子レベルの情報を提供する能力には限界があります。これに応えて、清華大学知能産業研究所の准研究員である周昊氏は、北京大学、南京大学、水木分子のチームと協力して、マルチスケールタンパク質言語モデルESM-AA(ESM All Atom)を提案し、残基拡張やマルチスケール位置エンコーディングなどのトレーニング メカニズムを設計することにより、原子スケールの情報を処理する能力が拡張されました。
ターゲットとリガンドの結合などのタスクにおける ESM-AA のパフォーマンスは大幅に向上し、ESM-2 などの現在の SOTA タンパク質言語モデルを上回り、Uni-Mol などの現在の SOTA 分子表現学習モデルも上回っています。関連する研究のタイトルは「ESM All-Atom: 統一分子モデリングのためのマルチスケールタンパク質言語モデル」です。機械学習に関するトップカンファレンスである ICML で発表されました。

用紙のアドレス:
https://icml.cc/virtual/2024/poster/35119
オープンソース プロジェクト「awesome-ai4s」は、100 を超える AI4S 論文の解釈をまとめ、大規模なデータ セットとツールを提供します。
https://github.com/hyperai/awesome-ai4s
データセット: タンパク質と分子データのハイブリッド データセットが構築されました
事前トレーニングタスクでは、この研究では、原子座標などの構造情報を含むタンパク質と分子のデータを組み合わせたデータセットを使用しました。
この研究では、タンパク質データセットとして、800 万個の信頼性の高い AlphaFold2 予測タンパク質配列と構造を含む AlphaFold DB を使用しました。
分子データセットについては、研究ではETKDGおよびMMFF分子力場によって生成された、1,900万個の分子と2億900万個の構成を含むデータを使用しました。
ESM-AAを訓練するとき、研究者らは最初にタンパク質データセットDpと分子データセットDmを最終データセットとして混合しました、つまりD=Dp∪Dm。原子のみからなる Dm の分子の場合、そのコード変換シーケンス X̄ は、残基のないすべての原子 Ā の順序集合、つまり R̄=∅ です。事前トレーニングでは分子データが使用されるため、ESM-AA はタンパク質と分子の両方を入力として受け入れることができることに注目してください。
ESM-AA モデルの構築: 統一された分子モデリングを実現するためのマルチスケールの事前トレーニングとエンコード
マルチ言語コード スイッチング手法からインスピレーションを得た ESM-AA は、予測およびタンパク質設計タスクを実行するときに、最初にいくつかの残基をランダムに解凍してマルチスケール コード スイッチング タンパク質 シーケンスを生成し、次に慎重に設計されたマルチスケール ポジション シーケンスを通じてこれらをエンコードします。訓練されており、その有効性は残基および原子スケールで実証されています。
タンパク質分子タスク、つまりタンパク質と低分子が関与するタスクを扱う場合、ESM-AA は追加のモデル支援を必要とせず、事前トレーニング済みモデルの機能を最大限に活用できます。
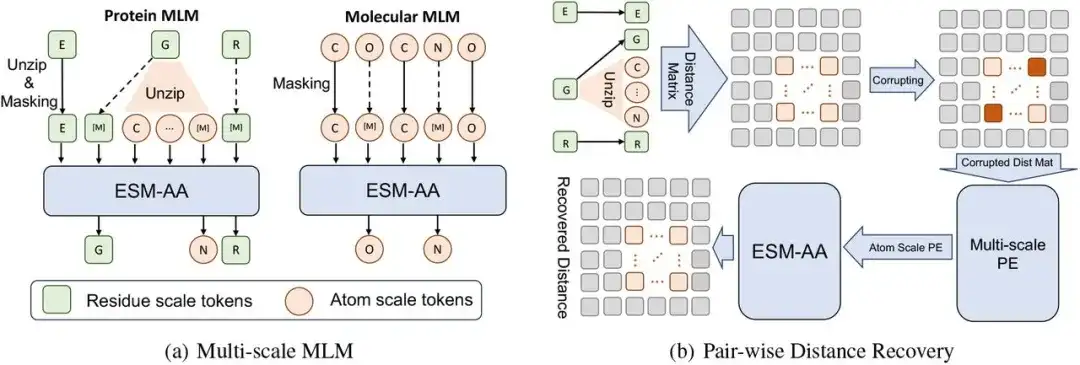
この研究のマルチスケール事前トレーニング フレームワークは、マルチスケール マスク言語モデル (MLM) とペアワイズ距離回復で構成されます。
具体的には、残基スケールでは、タンパク質 X は L 残基から構成される配列、つまり X = (r1,…,ri,…,rL) として見ることができます。各残基 ri は N 個の原子 A Ai={a1i,…,aNi} で構成されます。コードスイッチングタンパク質配列 X̅ を構築するために、研究では残基のセットをランダムに選択し、対応する原子を X に挿入する解凍プロセスを実装しました。このプロセスでは、研究者は解凍された原子を順番に配置し、最終的に原子セット Ai を X に挿入すると (つまり、剰余 ri を解凍すると)、コード スイッチング シーケンス X̄ が得られます。
続いて、研究者らは、コード切り替えシーケンス X̄ に対してマスクされた言語モデリングを実行しました。
まず、X̄ 内の原子または残基の一部がランダムに遮蔽され、モデルは周囲のコンテキストを使用して元の原子または残基を予測します。次に研究者らは、別の事前トレーニング タスクとしてデュアル ディスタンス リカバリー (PDR) を使用しました。つまり、原子スケールの構造情報は座標にノイズを追加することで破壊され、破壊された原子間距離情報がモデル入力として使用され、モデルはこれらの原子間の正確なユークリッド距離を復元する必要があります。
異なる残基にわたる長距離構造情報と単一残基内の原子スケールの構造情報との間の意味の違いを考慮して、この研究では残基内の PDR のみを計算します。これにより、ESM-AA は異なる残基内のさまざまな構造情報を学習することもできます。
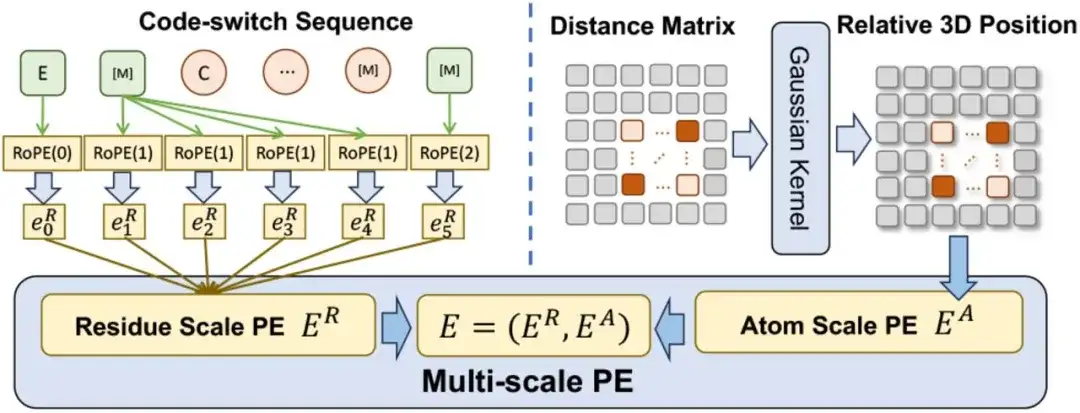
マルチスケール位置エンコーディング (Multi-scale Position Encoding) に関しては、研究者らはコード切り替えシーケンスにおける位置関係をエンコードするためにマルチスケール位置エンコーディング E を設計しました。 E には、残基スケールの位置コード ER と原子スケールの位置コード EA が含まれます。
ERの場合、研究者らは、純粋な残基配列を扱う際に元のエンコードとの一貫性を維持しながら、残基から原子への関係をエンコードできるように既存のエンコード方法を拡張しました。EAの場合、原子間の関係を捉えるために、この研究では空間距離行列を直接使用して原子の 3 次元位置をエンコードしました。
マルチスケール エンコード方式により、事前トレーニングがあいまいな位置関係の影響を受けないようにすることができ、ESM-AA が両方のスケールで効果的に機能できるようになることは言及する価値があります。
マルチスケール PE を Transformer に統合する際、この研究ではまず Transformer の正弦波エンコーディングを残差スケール位置エンコーディング ER に置き換え、原子スケール位置エンコーディング EA をセルフアテンションのバイアス値 (バイアス項) と見なしました。層。
調査結果: タンパク質の理解を最適化するための分子知識の融合
マルチスケールの統合事前トレーニング済みモデルの有効性を検証するために、この研究では、タンパク質や低分子を含むさまざまなタスクにおける ESM-AA のパフォーマンスを評価しました。
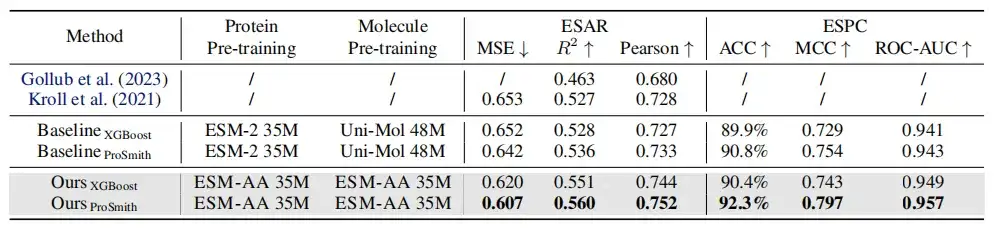
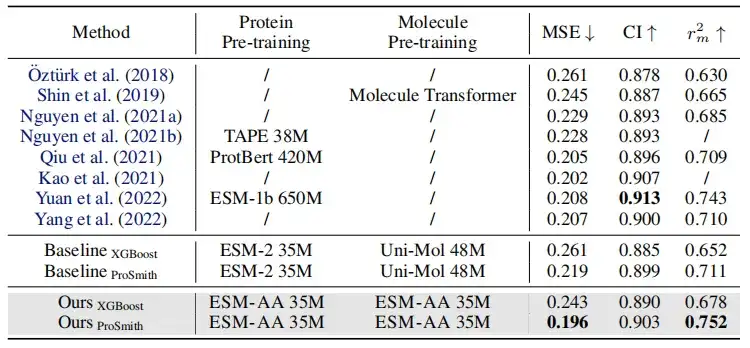
上の表に示すように、酵素-基質親和性回帰タスク、酵素-基質ペア分類タスク、薬物-標的親和性回帰タスクのパフォーマンス比較では、ほとんどの指標において、ESM-AA は他のモデルを上回り、最先端の結果を達成します。さらに、ESM-AA 上に構築された微調整戦略 (ProSmith や XGBoost など) は、2 つの独立した分子の事前トレーニング済みモデルとタンパク質の事前トレーニング済みモデルを組み合わせたバージョンよりも一貫して優れたパフォーマンスを発揮します (表 1 と表 1 および XGBoost の最後の 4 行に示すように)。 2)。
注目に値するのは、ESM-AA は、より大きなパラメータ サイズを持つ事前トレーニング済みモデルを使用するメソッドを無効にすることもできます(たとえば、表 2 の行 5、行 7、および最後の行の比較)。
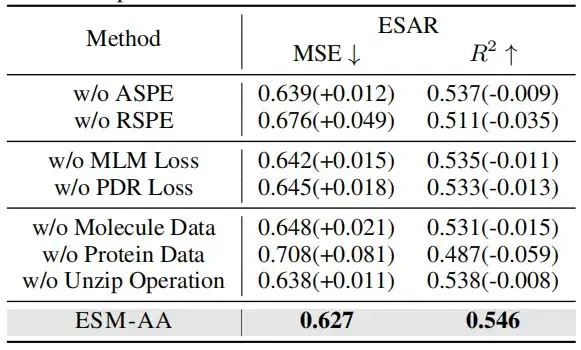
マルチスケール位置エンコーディングの有効性を検証するために、この研究では 2 つの条件下でアブレーション実験を実施しました。1 つは原子スケール位置エンコーディング (ASPE) を使用せず、もう 1 つは遺伝的スケール位置エンコーディング (RSPE) を使用しない状況で行われました。
分子またはタンパク質のデータが削除されると、モデルのパフォーマンスが大幅に低下しました。興味深いことに、タンパク質データを削除すると、分子データを削除するよりも顕著なパフォーマンスの低下が生じました。これは、モデルがタンパク質データでトレーニングされていない場合、タンパク質関連の知識がすぐに失われ、全体的なパフォーマンスが大幅に低下することを示しています。しかし、分子データがなくても、モデルは解凍操作を通じて原子レベルの情報を取得できます。
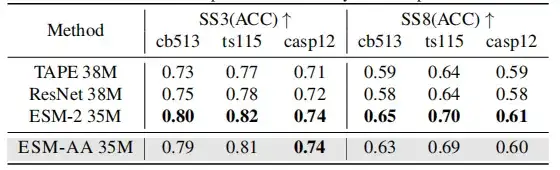
ESM-AA は既存の PLM に基づいて開発されたため、この研究では、二次構造予測タスクと教師なし接触予測タスクを使用して、タンパク質の事前トレーニング済みモデルの能力をテストすることで、タンパク質の包括的な理解を維持しているかどうかを判断することを目的としていました。構造の理解。
結果は、ESM-AA がこのタイプの研究では最適なパフォーマンスを達成できない可能性があることを示しています。ただし、二次構造予測や接触予測の性能は ESM-2 と同等です。
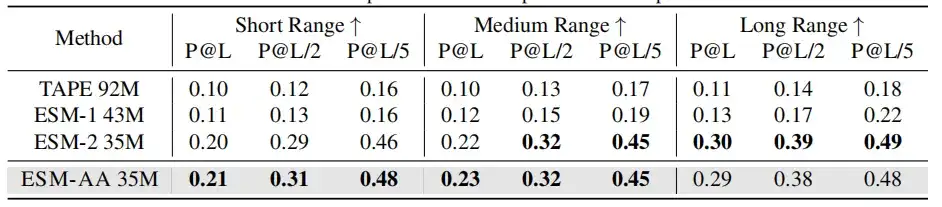
分子ベンチマークでは、ESM-AA は、ほとんどのタスクで Uni-Mol と同等のパフォーマンスを発揮します。そして多くの場合、いくつかの分子特異的モデルよりも優れたパフォーマンスを示し、分子タスクに対する強力なアプローチとしての出現を実証しています。
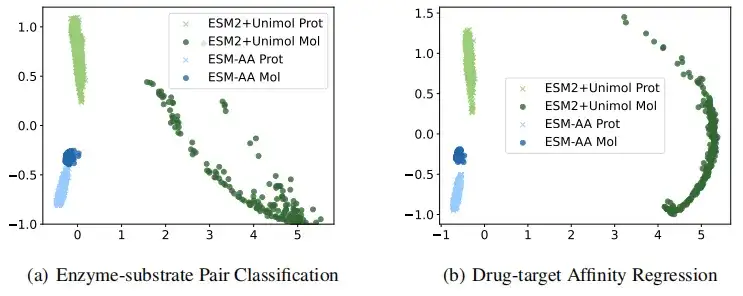
ESM-AA により高品質のタンパク質および小分子の特性評価が得られることをより直感的に説明するために、この研究では、酵素-基質ペア分類および薬物標的親和性回帰タスクにおいて ESM-AA と ESM-2+Uni-Mol を比較しました。抽出された表現は次のとおりです。視覚的に比較します。結果は次のようになります。ESM-AA モデルは、タンパク質と分子のデータを含む、より一貫性のある意味論的表現を作成できるため、ESM-AA は 2 つの個別の事前トレーニング済みモデルよりも優れています。
タンパク質言語モデル、大規模言語モデルの次の旅
1970 年代頃から、「21 世紀は生物学の世紀だ」と信じる科学者が増え、昨年 7 月、フォーブス誌は長い記事で、LLM が人々を生物学の分野に新たな時代に導いたと想像しました。変化の頂点。生物学は解読可能、プログラム可能、そしてある面ではデジタル システムであることが判明しました。LLM は、自然言語を制御する驚くべき能力により、生物学的言語を解読する可能性をもたらします。これにより、タンパク質言語モデルがこの時代で最も関心のある分野の 1 つになりました。
タンパク質言語モデルは、生物学における AI テクノロジーの最先端の応用を表しています。タンパク質の配列パターンや構造を学習することで、タンパク質の機能や形態を予測することができ、新薬の開発や病気の治療、基礎生物学研究などに大きな意義をもたらします。
これまで、ESM-2 や ESMFold などのタンパク質言語モデルは、AlphaFold に匹敵する精度を実証しており、より高速な処理速度と「オーファンタンパク質」のより正確な予測機能を備えています。これにより、タンパク質構造の予測が高速化されるだけでなく、タンパク質工学のための新しいツールが提供され、研究者が特定の機能を持つ新しいタンパク質配列を設計できるようになります。
さらに、タンパク質言語モデルの開発は、いわゆる「スケーリング則」の恩恵を受けます。つまり、モデルの規模、データセットのサイズ、計算量が増加するにつれて、モデルのパフォーマンスが大幅に向上します。これは、モデルパラメータの増加とトレーニングデータの蓄積により、タンパク質言語モデルの能力が質的に飛躍することを意味します。
過去 2 年間で、企業の世界でもタンパク質言語モデルが急速に発展する時期に入りました。 2023 年 7 月、Baitu Biosciences と清華大学は、最大 1,000 億 (100B) のパラメータ量を持つ xTrimo Protein General Language Model (xTrimoPGLM) と呼ばれるモデルを共同で提案しました。さまざまなタンパク質理解タスク (15 のタスクで大幅に使用されました) で使用されています。 13 のタスクにおいて他の最先端のベースライン モデルを上回ります)。生成タスクでは、xTrimoPGLM は天然のタンパク質構造に類似した新しいタンパク質配列を生成できます。
2024年6月、AIプロテイン企業Tushen Zhiheは次のように発表した。TourSynbio™ が開発した国内初の大規模自然言語タンパク質モデルを、すべての科学研究者および開発者にオープンソース化します。このモデルは、タンパク質の特性、機能予測、タンパク質の設計などの機能を含め、会話形式でタンパク質の文献を理解することを実現し、タンパク質の評価データセットを比較する評価指標においてはGPT4を超え、業界初となりました。
また、ESM-AAに代表される技術研究のブレークスルーは、技術開発が「ライト兄弟の瞬間」を超え、飛躍的な進歩を迎えようとしていることを意味しているのかもしれない。同時に、タンパク質言語モデルの応用は、医療やバイオ医薬品の分野に限定されず、農業、工業、材料科学、環境修復などの分野にも拡張され、これらの分野での技術革新を促進し、人類に起こる未曾有の変化。








