Command Palette
Search for a command to run...
1億個のパラメータを持つ大型セルモデルが登場! Nature サブジャーナルに掲載、清華大学チームが scFoundation: 20,000 個の遺伝子の同時モデリングを発表
近年、大規模な事前トレーニング済みモデルが人工知能の新たな波をリードしています。 「大規模モデル」は、大規模なマルチソース データから深層パターンを抽出することで、さまざまな分野のさまざまなタスクに対応するための「基本モデル」として機能します。たとえば、大規模な言語モデルは、大量のテキスト データを学習することで言語を理解し認識する能力を習得し、自然言語処理の分野のパラダイムに革命をもたらしました。
同様に、生命科学の分野では、生物も独自の「基礎的な言語」を持っています。細胞は人体の基本的な構造および機能単位です。DAN、RNA、タンパク質、遺伝子の発現値を「言葉」に例えると、それらの組み合わせが「細胞」という文を形成します。したがって、細胞の「言語」に基づいた大型の人工知能細胞モデルが開発できれば、生命科学や医学に新たな研究パラダイムや革新的な研究ツールが提供されることが期待されます。
しかし、現在、大規模な単一セル データのトレーニングには 3 つの主な課題があります。
* 遺伝子発現の事前トレーニング データは、さまざまな状態やタイプの細胞ランドスケープをカバーする必要があります。 現在、ほとんどの単一細胞 RNA シーケンス (scRNA-seq) データは大まかに編成されており、包括的で完全なデータベースはまだ不足しています。
* トレーニング プロセス中に、従来の Transformer では、約 20,000 個のタンパク質をコードする遺伝子で構成される「文」を処理するのが困難です。
*さまざまなテクノロジーや研究室からの ScRNA-seq データはシーケンシングの深さが異なるため、統一された意味のある細胞と遺伝子の表現をモデルが学習することができません。
これらの課題に対処するために、清華大学オートメーション学部基礎生活モデル研究室所長のZhang Xuegong教授、エレクトロニクス/AIR学部のMa Jianzhu教授、Baitu BiotechnologyのSong Le博士が協力して研究を実施しました。「単一細胞トランスクリプトミクスに関する大規模基礎モデル」と題された研究論文が、2024 年 6 月に Nature Methods に掲載されました。
この論文では、約 20,000 個の遺伝子を同時に処理できる scFoundation と呼ばれる大規模セル モデルが紹介されています。基本モデルとして、細胞配列決定の深さの強化、細胞の薬物応答予測、細胞の摂動予測など、さまざまな生物医学の下流タスクで優れたパフォーマンスの向上を示し、単一細胞研究における人工知能の新しいパラダイムを提供します。
研究のハイライト:
scFoundation cell の大規模モデルは、5,000 万個の細胞の遺伝子発現データに基づいてトレーニングされ、1 億個のパラメーターを持ち、約 20,000 個の遺伝子を同時に処理できます* このモデルは、計算とメモリの課題を軽減するために非対称設計を使用しています* このモデルは、遺伝子ネットワーク推論と転写因子の同定により新たな研究アイデアが得られる

用紙のアドレス:
https://www.nature.com/articles/s41592-024-02305-7
オープンソース プロジェクト「awesome-ai4s」は、100 を超える AI4S 論文の解釈をまとめており、大規模なデータ セットとツールも提供しています。
https://github.com/hyperai/awesome-ai4s
データセット: 包括的な単一セル データセットの構築
研究者らは、公的に利用可能なすべての単細胞リソースからデータを収集して、包括的な単細胞データセットを構築しました。これらには、遺伝子発現オムニバス (GEO)、単一細胞ポータル、HCA、ヒトゲノムプロジェクト (hECA)、深く統合されたヒト単一細胞オミックデータ (DISCO)、欧州分子生物学研究所 - 欧州バイオインフォマティクス研究所データベース (EMBL) - EBI) などが含まれます。
※GEOダウンロードアドレス:https://www.ncbi.nlm.nih.gov/geo/
*シングルセルポータルのダウンロードアドレス:https://singlecell.broadinstitute.org/single_cell
* HCA ダウンロード アドレス:https://data.humancellatlas.org/
* EMBL-EBI ダウンロードアドレス:https://www.ebi.ac.uk/
研究者らは、すべてのデータを、HUGO 遺伝子命名委員会によって特定された 19,264 個のタンパク質コード遺伝子および共通ミトコンドリア遺伝子の遺伝子リストと照合しました。データの品質管理を行った上で、5,000 万を超えるヒト scRNA-seq データが事前トレーニングのために取得されました。
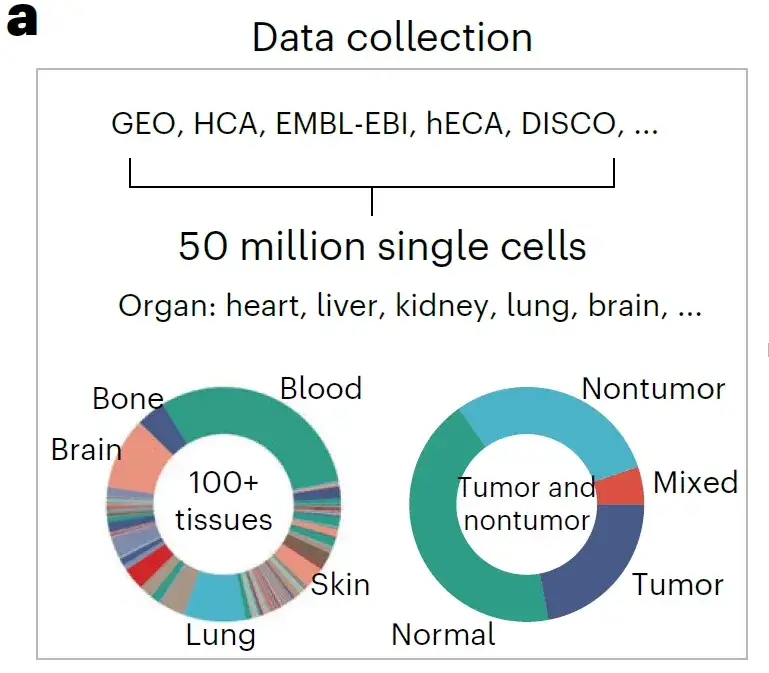
豊富なデータ ソースにより、生物学的パターンが豊富な事前トレーニング データセットが構築されます。解剖学的には、上に示したように、ほぼすべての既知のヒト細胞の種類と状態を含む、さまざまな疾患、腫瘍、正常な状態をカバーする 100 を超える組織タイプにまたがっています。
モデル アーキテクチャ: 1 億個のパラメーターを使用して scFoundation モデルを構築する
研究者らが開発したscFoundationモデルには約1億個のパラメータがあり、そのパラメータ規模、遺伝子網羅率、データ規模は単細胞分野で「最高レベル」にある。
モデル設計
研究者らは、scFoundation のバックボーン モデルとして xTrimoGene モデルを開発しました。これは、ベクトル モジュール (埋め込みモジュール) と非対称エンコーダ/デコーダ構造を含む、Transformer アーキテクチャに基づく拡張可能なモデルです。
その中で、ベクトル モジュールは、バニラ トランスフォーマー ブロックを使用して、エンコーダーが入力として非ゼロおよびマスクされていない発現遺伝子を受け取ることを保証するために、連続的な遺伝子発現スカラー値を学習可能な高次元ベクトルに変換します。デコーダはすべての遺伝子を入力として受け取り、パフォーマー ブロックを使用し、比較的少数のパラメータを持ちます。
この非対称設計により、他のアーキテクチャと比較して計算とメモリの課題が軽減されます。データは、同じパラメータ スケールを維持しながら、このモジュールで必要な計算量が従来の言語モデル Transformer の 3.4% のみであることを示しています。
トレーニング前のタスク
研究者らは、RDA (読み取り深度認識) モデリングと呼ばれる事前トレーニング タスクを設計しました。これは、大規模なデータにおけるシーケンスの深さの大きな分散を考慮した、マスクされた言語モデルの拡張です。
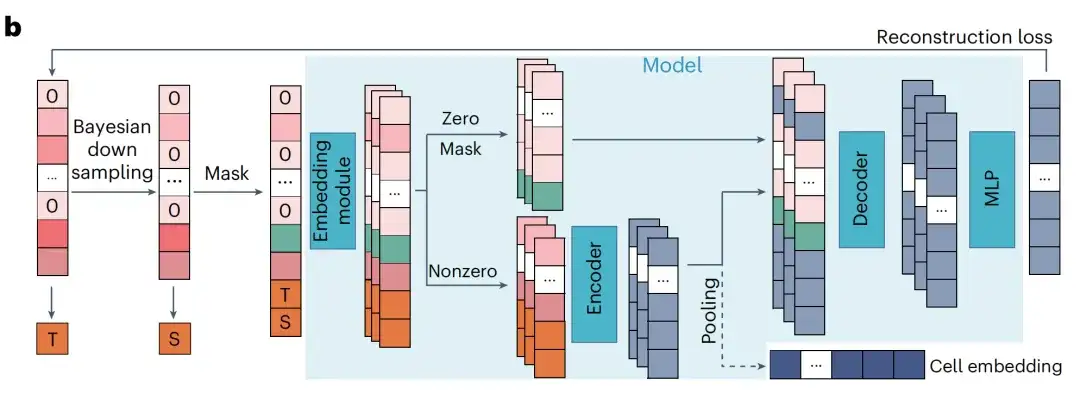
RDA モデリングでは、モデルは細胞のコンテキスト遺伝子に基づいてマスクされた遺伝子発現を予測します。研究者らは、総数を 1 つの細胞のシーケンス深度とみなし、2 つの総数メトリクス、T (ターゲット) と S (ソース) を定義しました。これらは、それぞれ元のサンプルと入力サンプルの総数を表します。研究者は、入力サンプル内のゼロ発現遺伝子と非ゼロ発現遺伝子をランダムにマスクし、それらのインデックスを記録します。
次にモデルは、マスクされた入力サンプルと 2 つのメトリクスを使用して、マスクされたインデックスでの元のサンプルの発現値を予測します。これにより、事前トレーニングされたモデルは、細胞内の遺伝子関係を捕捉するだけでなく、異なるシーケンス深度で細胞を調整することもできます。推論プロセス中に、研究者は細胞の生の遺伝子発現を事前学習済みモデルに入力し、T をその合計数 S よりも高く設定して、配列決定の深さを強化する遺伝子発現値を生成します。
簡単に言うと、RDA はシーケンス深度をダウンサンプリングできるため、トレーニング前の段階で従来のマスク回復タスクを完了することに加えて、モデルは低品質細胞から高品質細胞の遺伝子発現情報も回復できます。
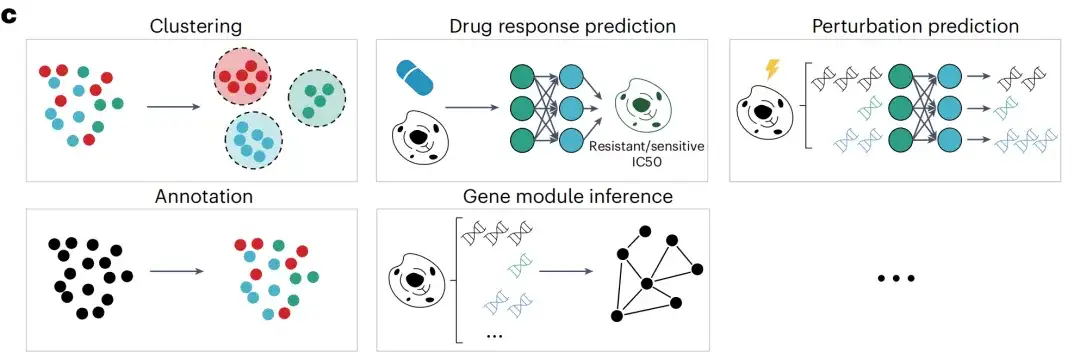
事前トレーニングが完了した後、研究者らはさらに scFoundation モデルを複数の下流タスクに適用しました。 scFoundation エンコーダーの出力は、クラスター化 (データセット内およびデータセット間)、バッチおよび単一細胞レベルの薬物反応予測、細胞型アノテーションなどの細胞レベルのタスクで使用するために細胞レベルのベクトルに要約されます。 scFoundation デコーダの出力は遺伝子レベルのコンテキスト ベクトルで、摂動予測や遺伝子モジュール推論などの遺伝子レベルのタスクに使用されます。
研究結果: scFoundation モデルは優れたパフォーマンスを実現
実際のアプリケーションでは、scFoundation モデルは「すぐに使用できる」モードと「微調整された」モードの 2 つのモードをサポートします。「すぐに使用できる」モードでは、独自の事前トレーニング タスクのおかげで、モデルを直接使用してセル データの品質を向上させ、追加の調整を行わずに既存のメソッドのパフォーマンスを達成または超えることができます。さらに、ユーザーは scFoundation を活用して、細胞の事前トレーニングされた表現を抽出できます。これは、細胞型固有の遺伝子モジュールと転写因子を特定するために使用でき、下流のタスクで広く使用できます。
スケーラブルで微調整不要のシーケンス深度強化モデル
研究者らは、それぞれ 3M、10M、100M のパラメーター サイズで 3 つのモデルをトレーニングし、検証データセットに損失を記録しました。
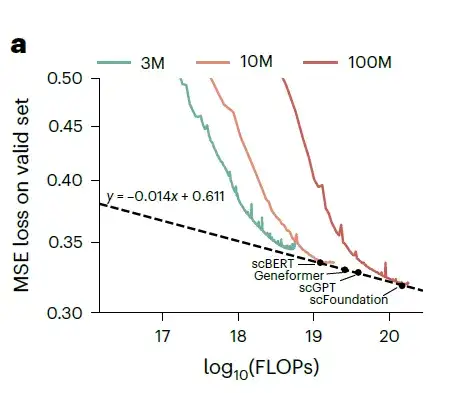
さまざまなパラメーター サイズと FLOP でのトレーニング損失。緑色の曲線は 3M モデルを表し、オレンジ色の曲線は 10M モデルを表し、赤色の曲線は 100M モデルを表します。
検証データセットの損失は、モデル パラメーターと浮動小数点演算 (FLOP) の数が増加するにつれて、べき乗則の減少を示しています。次に研究者らは、さまざまなサイズの xTrimoGene アーキテクチャ モデルのパフォーマンスを推定し、scVI と比較しました。上の図に示すように、1 億個のパラメーターを備えた scFoundation モデルは、すべてのモデルの中で最も優れたパフォーマンスを発揮しました。研究者らはさらに、細胞タイプの注釈タスクで 3 つのモデルを評価し、モデルのサイズが大きくなるにつれてパフォーマンスが向上することを観察しました。
研究者らは、検証データセットからランダムにサンプリングされた 10,000 個の細胞の独立したテストデータでこの機能を評価しました。このデータは、合計カウントを元のデータの 1%、5%、10%、および 20% にダウンサンプリングし、異なる合計カウント A データセットで 4 つのカウントを生成しました。さまざまな数字で。各データセットについて、微調整されていない scFoundation を使用して、予測値と実際の非ゼロ遺伝子発現の間の平均絶対誤差 (MAE)、平均相対誤差 (MRE)、およびピアソン相関係数 (PCC) を測定しました。
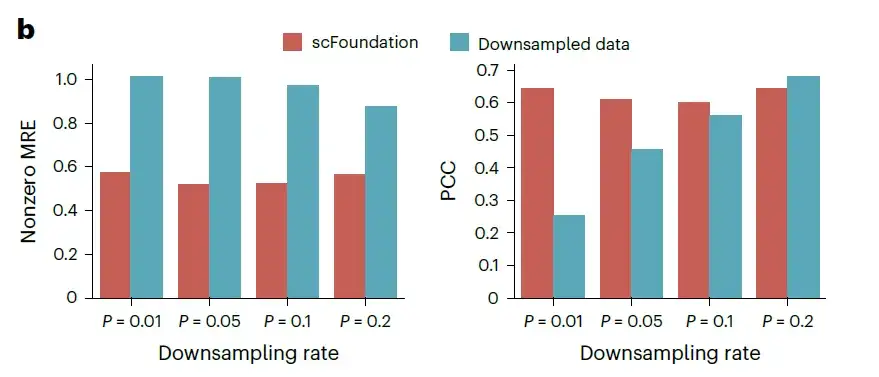
未知のデータセットに対するリード深度拡張パフォーマンスの評価 MRE と PCC を使用して、回復した遺伝子発現パフォーマンスを評価しました。MRE が低く、PCC が高いほど、パフォーマンスが優れていることを示します。
上の図に示されているように、scFoundation の MAE と MRE は、10% よりも低いダウンサンプリング レートでも大幅に半分に減少します。これらの結果は、scFoundation が非常に低い総数で遺伝子発現を増強できることを示しています。
下流タスク - 抗がん剤反応予測タスク
がん薬物反応 (CDR) は、薬物介入下での腫瘍細胞の反応を研究することを目的とし、CDR のコンピューターによる予測は、抗がん剤の設計を導き、がん生物学を理解するために重要です。この研究では、研究者らは scFoundation と CDR 予測手法 DeepCDR を組み合わせて、複数の細胞株データにおける薬物の半阻害濃度 IC50 値を予測し、scFoundation が単一細胞に基づく全体的な遺伝子発現データに有用な埋め込みを提供できるかどうかを検証しました。トレーニング情報。
研究者らは、複数の薬剤およびがん細胞株について、scFoundation ベースの結果と遺伝子発現ベースの結果を比較評価しました。結果は、ほとんどの薬剤とすべての種類のがんが、scFoundation の埋め込みを使用してより高いピアソン相関係数 (PCC) を達成したことを示しています。以下に示すように:
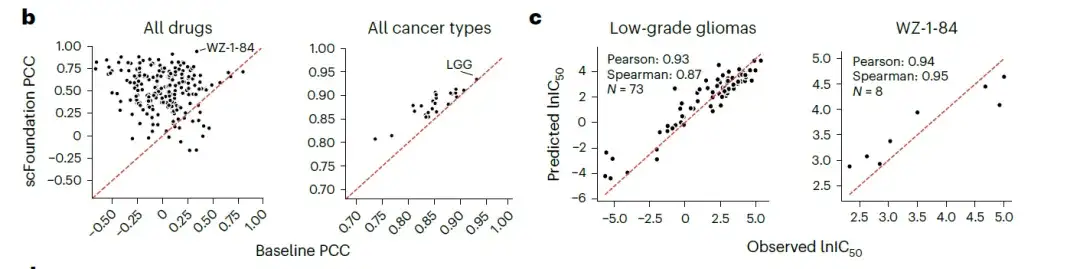
- 注: ピアソン相関係数は、変数間の線形関係の強さを測定する統計量であり、値の範囲は -1 から 1 です。相関係数が 1 に近い場合は、2 つの変数間に完全に正の線形関係があることを示し、-1 に近い場合は、完全に負の線形関係があることを示します。これは、2 つの変数間に線形関係がないことを意味します。
これは次のことを示していますscFoundation は単一細胞トランスクリプトーム データで事前トレーニングされていますが、学習された遺伝子関係はアンサンブル レベルの発現データに転送できます。圧縮ベクトルを生成して、より正確な IC50 予測を容易にします。したがって、scFoundation は、がん生物学における薬物反応の理解を拡大し、より効果的な抗がん治療の設計を導く確かな可能性を秘めています。
下流タスク - 単一細胞薬物反応分類タスク
単一細胞レベルで薬剤感受性を推測することは、異なる薬剤耐性プロファイルを示す特定の細胞サブタイプを特定するのに役立ち、それによって根底にあるメカニズムと新しい治療法についての貴重な洞察が得られます。したがって、研究者らは、SCAD と呼ばれる下流モデルに基づいて、単細胞薬物反応分類という重要なタスクに scFoundation を適用しました。
研究者らは、元の研究でより低いAUC(曲線下面積)値を示した4つの薬剤(ソラフェニブ、NVP-TAE684、PLX4720、およびエトポシド)に焦点を当てました。すべての遺伝子発現値を入力として使用して、scFoundation ベースのモデルとベースライン SCAD モデルを比較した結果、NVP-TAE684 とソラフェニブについて、scFoundation ベースのモデルがすべての薬剤の AUC 値でより高いスコアを達成したことが示されました。以下に示すように、AUC 値の増加は 0.2 を超え、特に顕著でした。
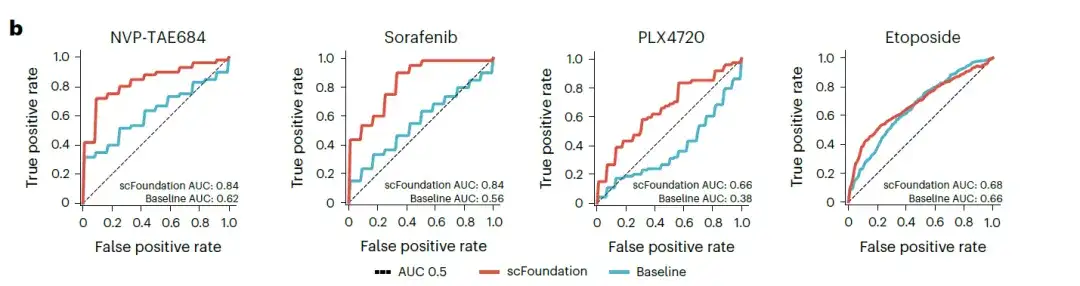
注: AUC はモデルのパフォーマンスを測定するために使用できます。AUC の値の範囲は 0 ~ 1 です。値が大きいほど、モデルの分類パフォーマンスが向上します。
これらの結果は、scFoundation 埋め込みを使用して薬剤感受性バイオマーカーシグナルを捕捉する可能性を検証します。
下流タスク - セル摂動予測タスク
摂動に対する細胞の反応を理解することは、生物医学への応用や医薬品設計にとって重要であり、異なる細胞型と潜在的な医薬品標的の間の遺伝子間相互作用を特定するのに役立ちます。研究者らは、scFoundation と高度なモデル GEARS を組み合わせて、単一細胞解像度で摂動応答を予測し、遺伝子前後で有意な差がある上位 20 個の差次的発現 (DE) 遺伝子の平均二乗誤差 (MSE) を計算しました。評価基準としての発現プロファイル。
結果は次のようになります。オリジナルの GEARS ベースライン モデルと比較して、scFoundation ベースのモデルはより低い MSE 値を達成しました。以下の図は、二重遺伝子摂動 ETS2 + CEBPE の上位 20 位の遺伝子発現変化を示しています。
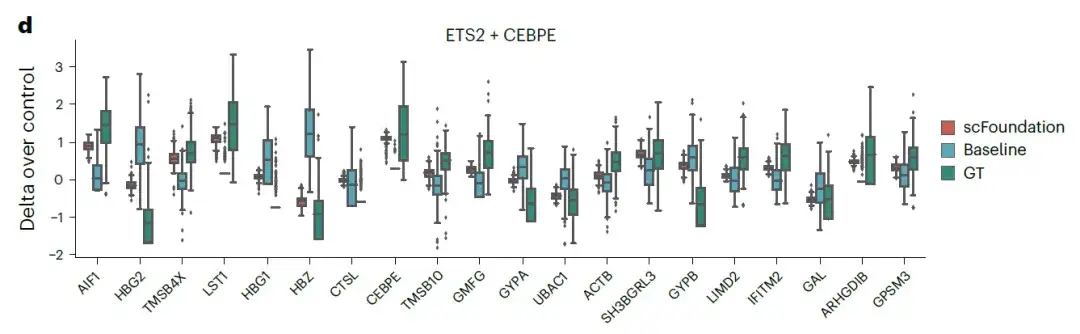
これらの結果は、個々の細胞の遺伝子表現を抽出して特定の遺伝子共発現ネットワークを構築することにより、scFoundation は、さまざまな条件下で細胞と遺伝子の表現を捕捉することに成功し、単一/二重摂動の予測精度を大幅に向上させました。
要約すると、scFoundation モデルは、モデル アーキテクチャ、トレーニング フレームワーク、大細胞事前トレーニング モデルの下流デモンストレーション アプリケーション システムを確立するための新しいアイデアと方法を提供し、生物医学タスクの学習のための基本機能を提供し、単一のトレーニングの範囲を拡大します。セル ドメインベースモデルの境界。
より優れたパフォーマンスを備えた大規模なライフ サイエンス モデルを探索する
世界有数の人工知能企業であるディープマインドの CEO 兼創設者であるデミス・ハサビス氏はかつて次のように述べています。「最も基本的なレベルでは、生物学は非常に複雑で動的な情報処理システムとみなすことができます。ちょうど数学が物理学の正しい記述言語であることが証明されたのと同じように、生物学は完璧な人工知能の応用分野になる可能性があります。」
ただし、従来の AI 手法では、正確な予測を行うために大量のラベル付きデータが必要です。しかし、ライフサイエンスでは、高品質のラベル付きデータが不足していることがよくあります。より少ないデータに基づいてより正確なダウンストリーム タスク モデルを構築したいということは、基礎となる基本モデルがより優れた表現または一般的な機能を備えている必要があることを意味します。したがって、生物学分野において、より優れた垂直大規模モデルの設計に取り組み始めている研究者が増えています。
2023 年 5 月トロント大学の研究チームは、単細胞生物学に基づいた初の大規模言語モデルである scGPT を発表しました。1,000 万個を超えるセルで事前トレーニングされたモデルにより、さまざまな下流タスクにわたる転移学習が可能になります。同年7月、チームはさらに3,300万個以上の細胞に対して事前トレーニングを生成することでscGPTの更新を試み、その結果、scGPTが遺伝子と細胞に関する重要な生物学的洞察を効果的に抽出し、それらをさまざまな下流タスクに実装できることが示されました。この機能には、マルチバッチ統合、マルチオミクス統合、細胞型アノテーション、遺伝的摂動予測、および遺伝子ネットワーク推論が含まれます。
この研究は「scGPT: 生成 AI を使用した単一細胞マルチオミクスの基礎モデルの構築に向けて」と題され、Nature Methods に掲載されました。
* 論文リンク:https://www.nature.com/articles/s41592-024-02201-0
2023 年 9 月中国科学院の多分野の研究チームで構成される「Xcompass Consortium」(Xcompassコンソーシアム)は、種を超えた生命の基盤を示す世界初の大規模モデル、GeneCompassの構築に成功した。このモデルは、ヒトとマウスの1億2,600万個以上の単一細胞のトランスクリプトームデータを統合し、プロモーター配列や遺伝子の共発現関係など4種類の事前知識を統合し、基本モデルパラメーター数は1億3,000万個に達し、遺伝子の制御を実現します。細胞の状態変化の予測とさまざまな生命プロセスの正確な分析をサポートしながら、規制法則を包括的に学習および理解します。

この研究は「GeneCompass: Knowledge-Informed Cross-Species Foundation Mode による普遍的な遺伝子制御メカニズムの解読」と題され、bioRxiv で公開されました。
2023 年 10 月、世界的な製薬大手サノフィは、BioMap との大規模な戦略的提携を発表し、両社は BioMap の最先端の発見モデルに基づいてバイオ治療薬を開発する予定です。
将来に目を向けると、人間の想像をはるかに超える複雑な理解能力とイノベーション創出能力を持つ、より複雑な生命の「自然言語」に大規模な言語モデルを適用することは、生命科学の研究パラダイムを真に変えることが期待されています。
参考文献:
1.https://www.jiqizhixin.com/articles/2023-9-29
2.https://www.tsinghua.edu.cn/info/1175/112118.htm
3.https://hope.huanqiu.com/article/4FYZxnpu88J
4.https://www.jiqizhixin.com/articles/2023-7-5-26'








