Command Palette
Search for a command to run...
がん細胞の増殖を抑制できる!恵湖薬科大学は天津医科大学と提携し、新しい腫瘍抑制タンパク質分解剤 dp53m を開発

知らない人も多いかも知れませんが、実際、私たち一人ひとりの中にがん細胞が存在します。人間の体は毎日、数十億、場合によっては数百億の細胞の再生と置換を行っていますが、この代謝プロセスでは、DNA 複製によって必然的に「エラー」が発生し、正常な細胞ががん細胞に変化することがあります。現場。しかし、人間の体内には腫瘍抑制タンパク質がいくつかありますが、p53 などのこれらのタンパク質は、細胞周期を調節することでがん細胞のアポトーシスや老化を促進し、それによってがんの発生を防ぐことができます。これは、私たちのほとんどががん細胞と「平和に共存」できる重要な理由でもあります。
「ゲノムの守護者」として知られる腫瘍抑制タンパク質 p53 は、遺伝子 TP53 によってコードされており、がんの予防に重要な役割を果たしています。ただし、TP53 は、R175、G245、R248、R273、R282 などのいくつかの一般的な特定のホット スポットでミスセンス変異を起こし、それによって p53 変異体が生成され、その結果、正常な腫瘍抑制タンパク質の機能が失われます。さらに、一部の p53 変異体のドミナント ネガティブ効果により、変異体 p53 は本来の腫瘍抑制因子機能を失うだけでなく、正常な野生型 p53 (p53-WT) の腫瘍抑制因子活性にも干渉し、その結果腫瘍の発生が増加します。リスク。
他のいくつかの p53 変異体と比較して、変異した p53-R175H タンパク質は、腫瘍形成、転移、薬剤耐性の可能性が高くなります。p53-R175H を標的とする薬剤を開発することにより、標的薬剤が p53-R175H を正確に識別して分解できるようにすることは、がんの発生を抑制する効果的な戦略となります。しかし、ほとんどの変異型 p53 タンパク質には小分子薬剤の活性部位が欠如しているため、変異型 p53 タンパク質を正確に認識して影響を与えることができる標的薬剤を設計することは非常に困難になります。
この点について、西安交通リバプール大学恵湖薬学部のウー・シジン教授、天津医科大学総合病院の謝松波教授と鍾典生教授のチーム、「p53-R175H ホットスポット変異誘発癌の正確な治療のための操作された DNA アプタマーベースの PROTAC」というタイトルの論文が Elsevier に掲載されました。
この研究では、選択的 p53-R175H デグレーダー -dp53m を開発しました。この分解剤は、変異型 p53-R175H タンパク質を特異的に認識することができ、細胞内の天然タンパク質分解システムであるユビキチン プロテアーゼ システムを利用して、標的タンパク質の標的分解を達成し、変異型 p53 タンパク質の機能発現を阻害します。
分解剤には顕著な抗腫瘍効果があり、明らかな毒性反応はありません。さらに、dp53m は化学療法薬シスプラチンと相乗作用して、がん治療に不可欠なシスプラチンに対するがん細胞の感受性を高めることもできます。
研究のハイライト:
- 研究者らは、MDシミュレーションのガイダンスの下で非コア塩基を修飾し、反復分子ドッキングガイドポストSELEX法によってRNAアプタマーをDNAアプタマー(p53m-DA)に誘導した。
- dp53m の構成要素として、p53m-DA は p53-R175H タンパク質を特異的に認識でき、CRBN はタンパク質の分解プロセスに関与するため、dp53m は p53-R175H タンパク質を特異的に認識して分解できます。

用紙のアドレス:
https://pubmed.ncbi.nlm.nih.gov/38811338
オープンソース プロジェクト「awesome-ai4s」は、100 を超える AI4S 論文の解釈をまとめており、大規模なデータ セットとツールも提供しています。
https://github.com/hyperai/awesome-ai4s
Post-SELEXエンジニアリング:変異型p53-R175Hを特異的に認識できる高性能DNAアプタマーの構築
p53の研究において、研究者らは、R175H変異がp53の構造状態を変化させることによってDNA結合機能に影響を及ぼし、さまざまな阻害剤小分子とアプタマーの組み合わせによってp53-R175Hの活性を部分的に回復させることができるが、その影響は分子に影響を与えることを発見した。メカニズムは報告されていない。
この研究では、最初に分子シミュレーション法を使用して、p53-R175H の機能を回復するアプタマーの固有の分子機構を調査しました。正常なp53-WTと比較して、変異体p53-R175Hでは、L3とCヘリックスの間の距離が近く、L2とL3の間の距離が大きくなっていることが、変異体p53の発生に関与している可能性があります。 -R175H。その典型的な機能を失う主な理由は、そのアプタマーおよび関連する PROTAC 分子の特異的認識の構造的基盤も提供します。
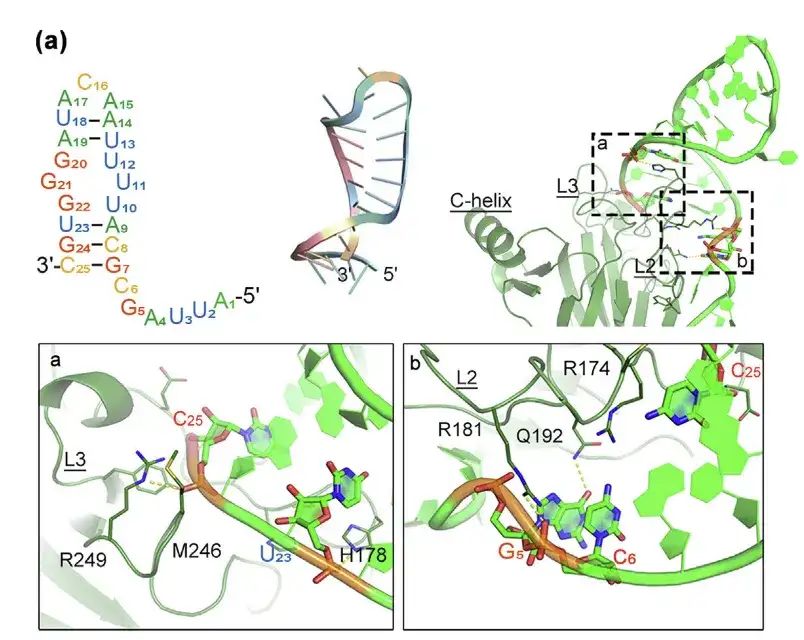
- 分子シミュレーション手法を用いたp53m-RAの構造およびp53-R175Hタンパク質との相互作用の予測
- 結合されたインターフェイスは領域 a と b に表示されます。
これまでの研究により、p53-R175Hを標的とするRNAアプタマー(p53m-RA)はp53-R175Hに対して高い親和性を有することが確認されていたが、血清中では不安定であるため実用化には限界があった。
参照元:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9884801
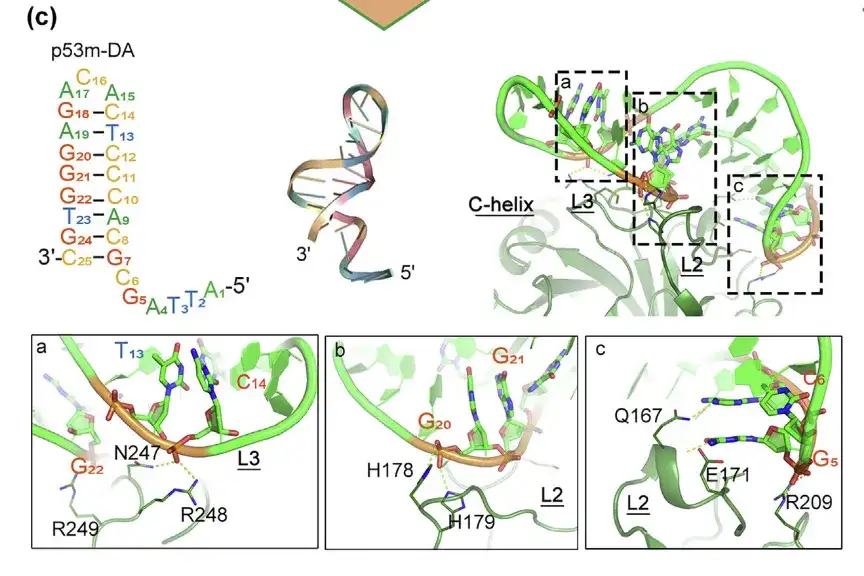
SELEX 後に最適化された p53m-DA とその p53-R175H との相互作用
結合インターフェースはエリア a、b、c にあります
アプタマーの構造安定性と親和性を高めるために、研究者らはMDシミュレーションの指導の下で非コア塩基を修飾し、ポストSELEXを通じてRNAアプタマーをDNAアプタマー(p53m-DA)に操作した。
この研究における分子シミュレーションの計算能力は、 オープンベイズ 供給
結果は、p53m-DAのペア領域の構造が主溝として現れ、不対ループ領域が副溝であることを示しています。主溝は L2 の H178、H179、E171、Q167 との結合に重要な役割を果たし、副溝は L3 の N247、R248、R249 との相互作用に重要な役割を果たします。さらに、p53m-DA はシミュレーション全体を通じて二本鎖らせん構造を維持し続け、その構造の安定性を示しました。
L2とL3は変異タンパク質に存在する領域です
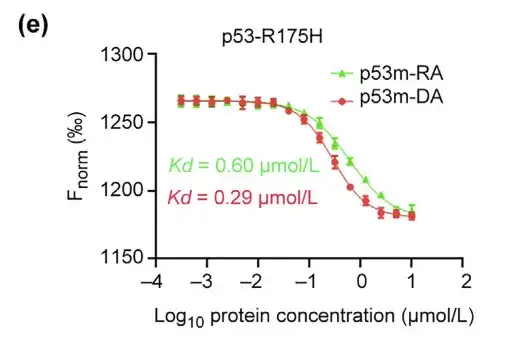
研究者らはさらに、p53-R175H に対する p53m-DA の親和性と選択性を評価しました。 MST アッセイにより、p53m-DA の Kd は 0.29 lmol/L であり、p53m-RA よりも約 2 倍低いことが示されました。
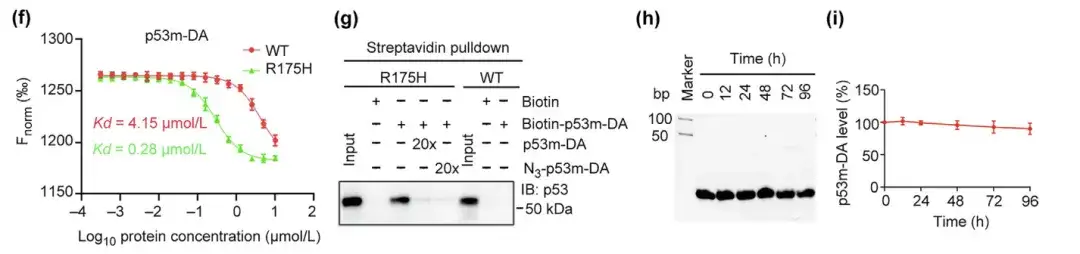
(f) p53-WT および p53-R175H に対する p53m-DA の親和性
(g) p53-R175H への p53m-DA の特異的結合を分析するストレプトアビジン プルダウン アッセイ
(h,i) p53m-DA の血清安定性を評価し、p53m-DA の残存量を決定する
さらに、変異型 p53-R175H に対する p53m-DA の親和性は、正常な p53-WT に対するその親和性よりも 14 倍高かった。ストレプトアビジンプルダウンアッセイでは、p53m-DA は p53-WT と相互作用することなく p53-R175H に特異的に結合し、p53m-DA の p53-R175H に対する特異性を示しました。注目すべきことに、p53m-DA の血清安定性は大幅に改善され、96 時間後の分解は最小限でした。
総括する、p53m-DAは、高い特異性と安定性を備えたアプタマーです。
dp53m: 特異的な p53-R175H 分解剤です
実験 1: dp53m の合成
アプタマー p53m-DA の 5' 末端は、p53-R175H との結合部位の外側に位置しています。研究者らは、クリック反応を使用して、アルキニル化サリドマイド (CRBN E3 リガーゼのリガンド) を p53m-DA の 5' 末端の N3 に結合させました。 、結果として生じるタンパク質分解を標的とするキメラPROTAC(dp53mと命名)は、p53-R175 Hに選択的に結合することができる。 dp53m の構造を以下の図に示します。
*PROTAC: E3 リガーゼ リガンド、POI ターゲティング リガンド、およびリガンド間の化学リンカーで構成されます。
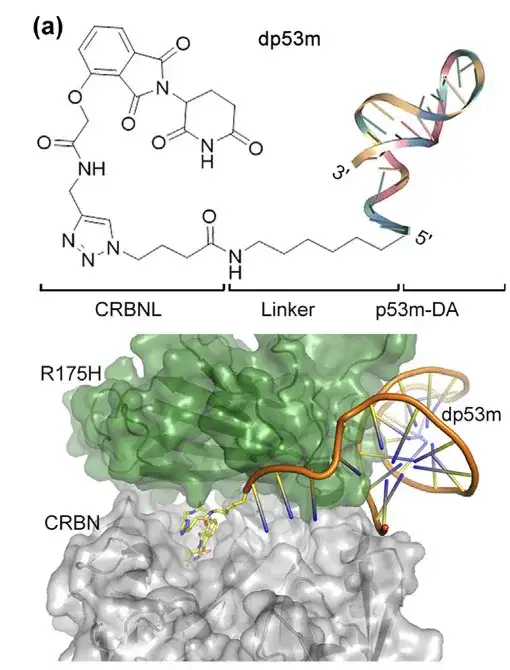
(a) タンパク質間ドッキングと分子動力学計算を用いた dp53m の構造シミュレーション
下: p53-R175H および CRBN との結合界面
実験 2: dp53m は変異体 p53-R175H を特異的に認識して分解することができる
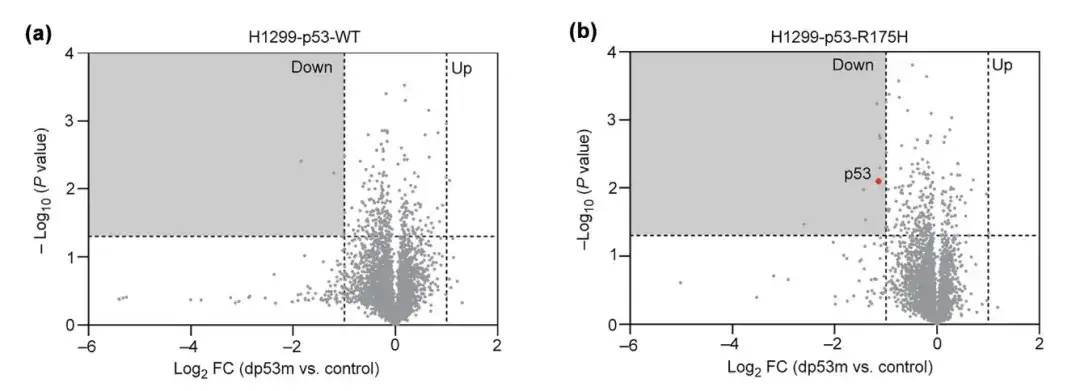
p53-R175Hに対するdp53mの細胞特異性を研究するために、研究者らは、H1299細胞で過剰発現したp53-R175Hおよびp53-WTをPBSまたはdp53mで16時間処理し、それらのタンパク質変化レベルをモニタリングした。結果は、dp53m は p53-R175H を有意に分解するが、p53-WT は分解しないことを示しました。
*PBS はリン酸緩衝生理食塩水であり、対照群として使用されました。
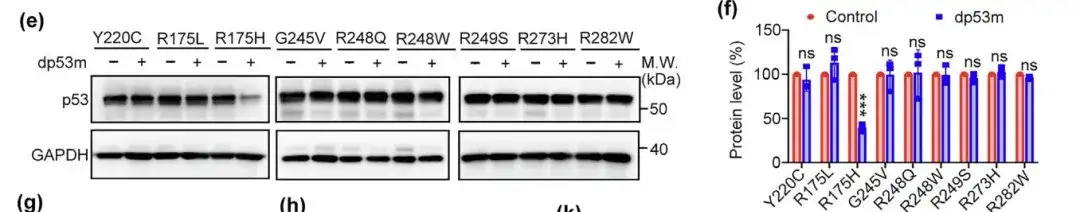
さらに研究者らは、Y220C、R175L、G245V、R248Q、R248W、R249S、R273H、R282Wなどの他のホットスポット変異体に対するdp53mの分解能力も評価した。結果は、dp53m が p53-R175H のみを効果的に分解することを示しています。p53-R175H 分解に対するその特異性が実証されました。
p53WT と比較して、dp53m 処理は p53-R175H のポリユビキチン化を増加させました。また、dp53m の分解はプロテアーゼ阻害剤 MG132、CRBN リガンド、または CRBN 標的 siRNA によってブロックできることが研究で判明しているため、dp53m による p53-R175H の分解は、ユビキチン - プロテアソーム機構を通じて起こると推測されます。
総括する、dp53m は特異的な p53-R175H 分解物質であり、その分解プロセスはユビキチン - プロテアソーム機構を通じて起こります。
dp53m はがん細胞の増殖と遊走、および腫瘍の成長を阻害します
実験 1: dp53m は、p53-R175H によって引き起こされる癌細胞の増殖と移動を阻害します。
研究者らは、H1299-p53-WT、H1299-p53-R175H、A549、デトロイト 562、SKBR3 細胞をそれぞれ PBS、p53m-DA、または dp53m で処理し、生存率アッセイを行って細胞内のコロニー形成を観察しました。
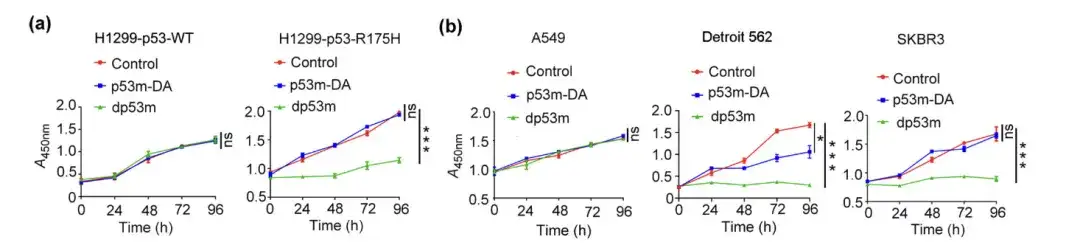
PBS、p53m-DA、または dp53m で処理した H1299-p53-WT、H1299-p53-R175H、A549、Detroit 562、および SKBR3 細胞の生存率アッセイ
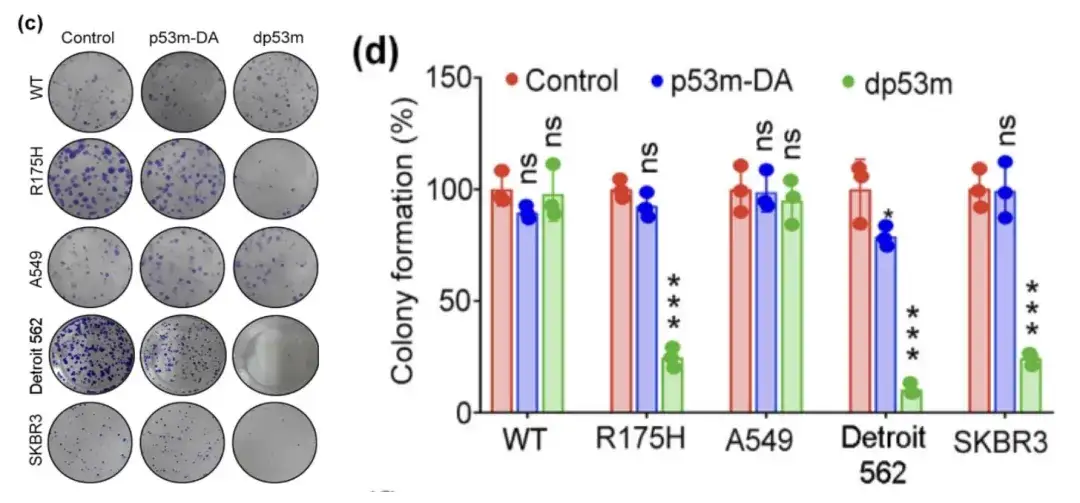
* dp53m は、H1299-p53-R175H、Detroit 562、および SKBR3 細胞におけるコロニー形成を阻害します。
*H1299-p53-WT および A549 細胞では有意な効果は観察されませんでした
上の図は、dp53m が p53-R175H (H1299-p53-R175H、Detroit 562、SKBR3) を発現する癌細胞では増殖に対して顕著な抑制効果を示すのに対し、p53-WT を発現する細胞では基本的に影響を受けないことを示しています。これは、dp53m が p53-R175H 変異を持つ癌細胞において優れた治療可能性を持っていることを示唆しています。
実験 2: dp53m は in vivo で p53-R175H によって引き起こされる腫瘍増殖を阻害します
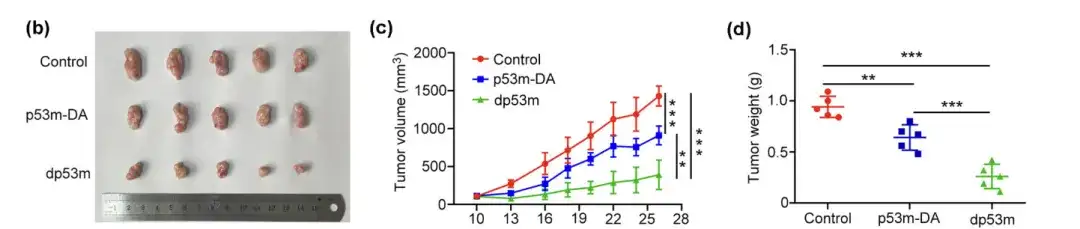
(b) 治療後の孤立腫瘍の肉眼的外観
(c) 示された時間に測定された腫瘍体積。
(d) 最終的な腫瘍重量の決定
研究者らは、腫瘍を有するBALB/cマウスを3つのグループにランダムに割り当て、対照としてp53m-DA、dp53m、または生理食塩水を静脈内投与し、対照群またはp53m-DAと比較した。dp53m は腫瘍の増殖を有意に阻害しました。
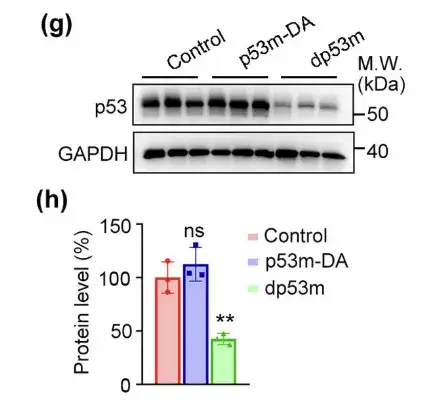
予想通り、dp53m は、腫瘍における p53-R175H の発現レベルを効果的に低下させました。
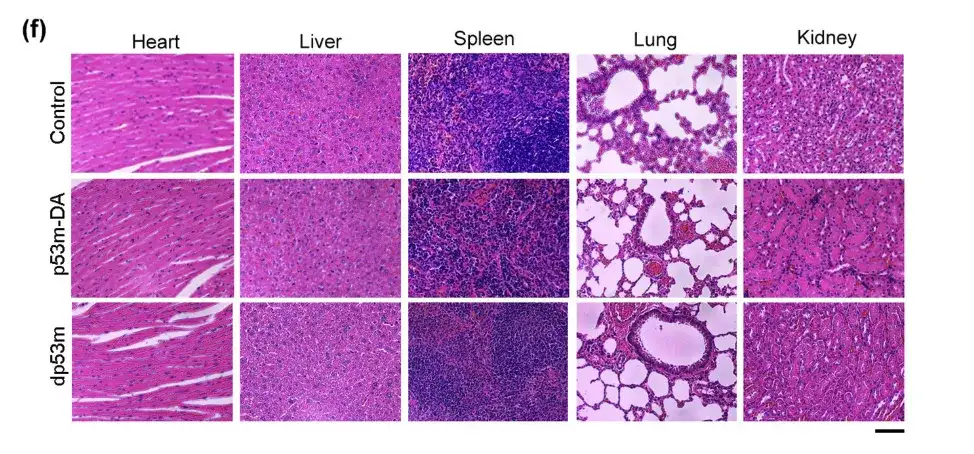
上記では、3 つのマウス グループ間で、心臓、肝臓、肺、脾臓、腎臓組織の組織学に有意な差はありません。これは、dp53m による毒性が生体内では存在しないことを示しています。
総括する、dp53m は、明らかな毒性作用を伴わずに、p53-R175H によって引き起こされる腫瘍増殖を阻害します。
さらに、変異p53-R175Hがシスプラチンなどの化学療法薬に対する耐性と関連していることを考慮して、研究者らはシスプラチンの抗腫瘍活性に対するdp53mの影響も分析した。結果は、dp53m とシスプラチンの間に強力な相乗相互作用があることを示しています。つまり、dp53m は、シスプラチンに対する p53-R175H 癌細胞の感受性を高めることができます。がん治療における「無敵のパートナー」となります。
抗がん防御ラインを強化し、科学者たちが協力して希望の道を築く
制御されない細胞増殖によって引き起こされる病気であるがんの発生率は、世界的な高齢化、環境汚染の増加、生活習慣の変化に伴い大幅に増加しています。国際がん研究機関 (IARC) のデータによると、世界の新規がん症例数は 2020 年の約 1,929 万件から 2040 年には約 3,023 万件に、56.7% 増加すると推定されています。
がんとの世界的な闘いにおいて、研究者は重要な役割を果たしています。この研究の責任著者は次の 3 名です。西安交通リバプール大学恵湖薬学部のウー・シジン先生、天津医科大学総合病院の謝ソンボ教授、ゾン・ディアンシェン教授は、この分野の優れた代表者です。
その中で、ウー・シジン教授は、新しい標的薬物の研究開発に焦点を当てており、現在の研究の焦点は、モデリング、ファーマコフォア解析、ドッキング、分子動力学などのシミュレーション手法を使用した、コンピューター支援による薬物の設計と発見です。 MD) 新しい治療薬を特定する。同氏は2022年の研究で、SOST発現の上方制御が乳がん患者の予後不良と関連しており、SOSTが下流のシグナル伝達経路を活性化して乳がん細胞の増殖と骨転移を促進することを発見した。この研究では、コンピューターによるスクリーニングを通じて、SOSTとSTAT3の間の相互作用を阻害してSTAT3のリン酸化を阻害し、乳がんの骨転移を軽減する候補治療化合物S6を同定した。
用紙のアドレス:
https://pubmed.ncbi.nlm.nih.gov/36581888
さらに、Xie Songbo教授の主な研究方向は、標的タンパク質の分解と薬物送達であり、2023年の研究では、アプタマーを「標的弾頭」として使用することで、「不可能な薬用タンパク質の分解」を誘導するための新しい戦略が使用できると提案した。この概念を確認するために、研究者らは標的として発がん性ヌクレオリン(NCL)を選択し、最終的にユビキチンプロテアーゼシステム依存的にNCL分解を誘導し、NCL誘導性の乳がん細胞増殖を阻害できる一連のNCL分解因子を生成した。 、この研究はタンパク質分解に関する新たな視点を提供するだけでなく、癌などの疾患の治療薬開発のための強固な基盤を築きます。
用紙のアドレス:
https://pubmed.ncbi.nlm.nih.gov/36608275
最後に、Zhong Diansheng 教授は、肺がんの早期診断、化学療法、標的療法、抗血管新生療法の分野でも詳細な研究を行っており、90 以上の学術論文を発表しています。同氏は2024年の研究で、CBX4がPHGDH発現とセリン生合成を上方制御することで肺腺がん(LUAD)の増殖を促進し、同時にZEB2転写を阻害することでLUADの転移を阻害することを発見した。この発見は、CBX4 とエピジェネティック因子の間の相互作用を理解するのに役立ち、LUAD の潜在的な治療経路への洞察を提供します。
用紙のアドレス:
https://www.nature.com/articles/s41419-024-06745-z
ウー・シジン教授、謝松博教授、鍾典生教授以外にも、黙々と献身的に努力している多くの科学者や医師がいます。私たちは、科学技術の絶え間ない進歩により、いつか癌がなくなることを願っています。人類にとっては悪夢。








