Command Palette
Search for a command to run...
独自の研究開発!軍事医学研究所チームは、単一細胞マルチオミクスデータのモザイク統合に使用できる MIDAS を提案しました
ご存知のとおり、細胞は生命の最小単位であり、私たちの成長と発達の基礎となる細胞は 40 ~ 60 兆個あります。細胞だけでなく病気の診断や治療も重要です。
近年、単一細胞シーケンシング技術が突然出現し、分子生物学研究のホットスポットとなっています。業界では、疾患や発生などの臨床および基礎研究課題に焦点を当てた大量の単一細胞シーケンシングデータが生成されています。しかし、さまざまなオミクスの組み合わせ、さまざまなシーケンス技術、さまざまなシーケンスサンプルから得られた膨大なデータは、床のモザイクタイルと同じくらい分散していて多様です。このような膨大で乱雑なデータをどのように統合して生物医学研究を実施するかは、世界中の科学者が直面する共通の課題です。
この課題を克服するために、最近、軍事医学科学アカデミーのイン・シャオミン氏のチームとボー・シャオチェン氏のチームは、 ネイチャーバイオテクノロジー このジャーナルは、「MIDAS を使用した単一細胞マルチモーダル データのモザイク統合と知識伝達」というタイトルの研究論文を掲載しました。この研究では、単一細胞マルチモーダル オミクス (scMulti-omics) データ モザイク統合 (つまり、異なるデータ セットが一部の検出モダリティのみを共有する) と知識伝達のためのコンピューティング ツール MIDAS を提案します。自己教師あり学習と情報理論的アプローチに基づいて、単一細胞マルチオミックスモザイクデータのモーダルアライメント、データ補完、およびバッチ補正の一般的な統合機能が初めて実現され、大規模なシステムを構築するための基盤が提供されます。大規模マルチオミクス細胞アトラス、大規模単一細胞マルチオミクス解析および知識伝達は、重要な独自技術を提供します。
研究のハイライト:
* 生成人工知能に基づいた新しいアルゴリズム MIDAS を独自に開発
* モーダルアライメント、データ補完、単一細胞マルチオミクスモザイクデータの一括補正などの一般的な統合機能が初めて実装されました。
※この新しいアルゴリズムは、細胞の機能や分子調節機構の解明、疾患の発生や発症の研究にとって極めて重要です。

用紙のアドレス:
https://www.nature.com/articles/s41587-023-02040-y
公式アカウントをフォローし、バックグラウンドで「Single Cell」に返信すると全文PDFが入手できます
データセット: 複数のデータセット、多次元の評価パフォーマンス
MIDAS モデルの利点をさまざまな側面から比較するために、この研究では複数のデータセットを構築しました。
まず、MIDAS と最先端の手法を比較します。この研究では、完全なモダリティを使用した三峰性統合 (モザイク統合の簡略化された形式) における MIDAS のパフォーマンスを評価します。研究チームは、このタスクを「長方形統合」と名付けました。チームは、公開されている 2 台の単セル三脚人間を使用します PBMC 各細胞のデータセット(DOGMA-seqおよびTEA-seq)、RNA、ADTおよびATACを同時に測定し、それによってdogma-fullデータセットおよびteadog-fullデータセットを構築した。注: PBMC は末梢血単核球の略で、免疫学の分野の科学研究活動で一般的に使用されます。
次に、テッセレーション統合における MIDAS のパフォーマンスを評価するために、研究チームはさらに、以前に生成された長方形データセットに基づいて 14 個の不完全なデータセットを構築しました。各モザイク データセットは、フルモーダル データセットから複数のモーダル バッチ ブロックを削除することによって生成されました。
第三に、MIDAS の知識伝達能力を研究するために、研究チームは、アトラス データ セットをアトラス構築用の参照データ セットとクエリ データ セットに再分割しました。研究チームは、アトラスから DOGMA-seq を削除することで、atlas-no_dogma という名前の参照データセットを取得しました。
4 番目に、連続的なセル状態変化を伴う単一セル データセットにおける MIDAS の適用を研究するため、研究チームは、公開されている scRNA-seq (単一細胞 RNA シーケンス) から取得した 3 つの異なるサンプル (ICA、ASAP、CITE) を組み合わせて、ヒト BMMC モザイク データセットを構築しました。
モデルアーキテクチャ: ディープ生成モデル MIDAS
MIDAS は、トランスポザーゼ アクセス クロマチン (ATAC)、RNA、および抗体由来タグ (ADT) の測定を組み込んだ、不完全な単一細胞マルチモーダル データの結合分布を表す深層生成モデルです。
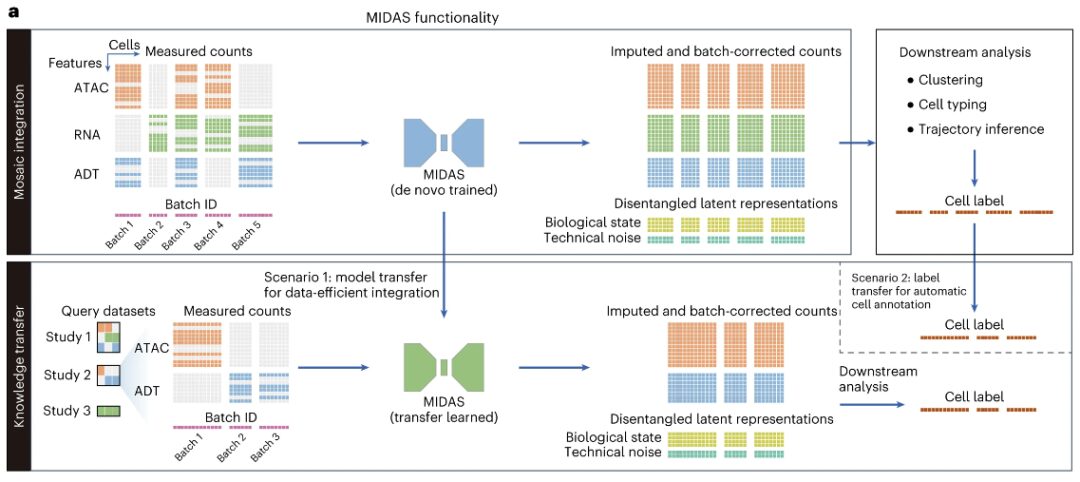
具体的には、MIDAS は、ディープ ニューラル ネットワークに基づいて、各セルのマルチモーダル測定が、モダリティに依存せず分離された 2 つの潜在変数 (生物学的状態と技術的ノイズ) を通じて生成されることを前提としています。その入力には、さまざまな単一セル サンプル (バッチ) で構成されるモザイク特徴セル数行列と、セル バッチ ID を表すベクトルが含まれます。これらの単一細胞サンプルは、異なる実験に由来するか、異なるシーケンス技術 (scRNA-seq、CITE-seq、ASAP-seq、TEA-seq など) を適用することによって生成されるため、技術的なノイズ、モダリティ、特性が異なる場合があります。 。
MIDAS の出力には、生物学的状態および技術的ノイズ行列に加えて、入力データ内の欠落したモードと特徴が補間され、バッチ効果が除去される、代入およびバッチ補正されたカウント行列が含まれます。これらの出力は、クラスタリング、細胞タイプの描写、軌跡推論などの下流分析に使用できます。
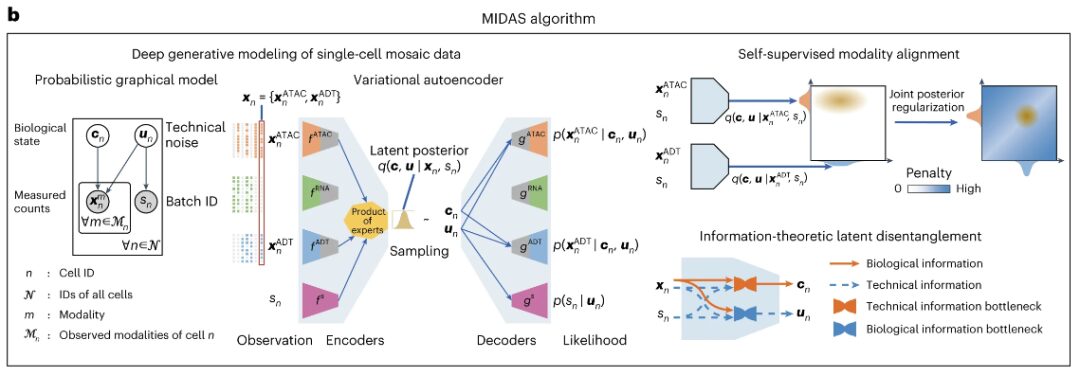
MIDAS は変分オートエンコーダ (VAE) アーキテクチャに基づいており、モジュール式エンコーダ ネットワークとデコーダ ネットワークを備えています。前者はモザイク入力データを処理して潜在変数を推論でき、後者は潜在変数を使用して観測データの生成を開始できます。 。 MIDAS は、自己教師あり学習を使用して潜在空間内のさまざまなモダリティを調整し、補間や翻訳などの下流タスクにおけるクロスモーダル推論を向上させます。同時に、情報理論手法も適用されて生物学的状態と技術的ノイズを分離し、バッチ補正をさらに実現します。
研究者らは、これらの要素をこの研究の最適化目標に組み合わせ、確率的勾配変分ベイズ (SGVB) を通じて MIDAS のスケーラブルな学習と推論を実現しました。これにより、単一セルのマルチモーダル データの分析も可能になりました。可能。さらに、構築されたマップ内の知識をさまざまなモードの組み合わせでクエリ データセットに転送するために、研究者らは、モデル パラメーターとセル ラベルをそれぞれ転送するための転移学習スキームと相互参照マッピング スキームを開発しました。
研究結果: MIDAS は多用途かつ効率的です
研究結果は、MIDAS が強力で多用途かつ効率的な単一セルのマルチモーダル統合ツールであることを示しています。
バッチ効果の除去と生物学的シグナルの保存の観点から、研究チームは MIDAS のパフォーマンスを最近発表された 9 つの方法と比較しました。
結果は次のことを示していますMIDAS はバッチ効果を理想的に排除し、dogma-full および teadog-full データセットのセルタイプ情報を保持しますが、他の方法のパフォーマンスは若干劣ります。たとえば、BBKNN+average、MOFA+、PCA+WNN、Scanorama-embed+WNN、Scanorama-feat+WNN は異なるバッチをうまく混合せず、PCA+WNN と Scanorama-feat+WNN はセルを含む非常に大きなセル クラスターを生成します。程度が一貫していないタイプ。
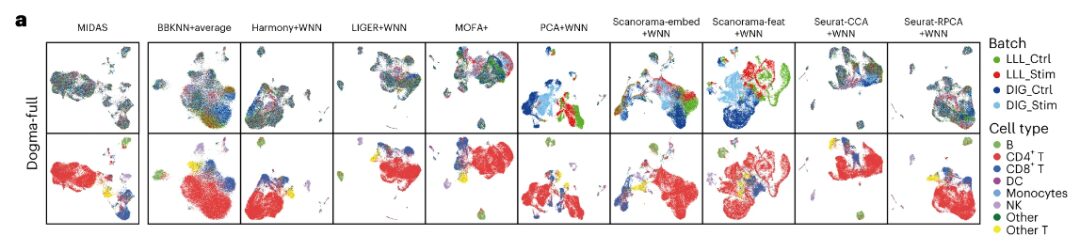
評価結果と下流分析結果の取得
バッチの配置に関して - MIDAS は、セルの異なるバッチを配置し、セル タイプ ラベルを使用してそれらを一貫してグループ化するという優れた仕事をします。他の方法では、細胞の異なるバッチをうまく混合できず、細胞の種類と大きく一致しない細胞クラスターが生成されます。 scIB ベンチマークは、MIDAS がさまざまなテッセレーション タスクで安定したパフォーマンスを示し、全体的なスコアが他の方法よりもはるかに高いことを示しています。
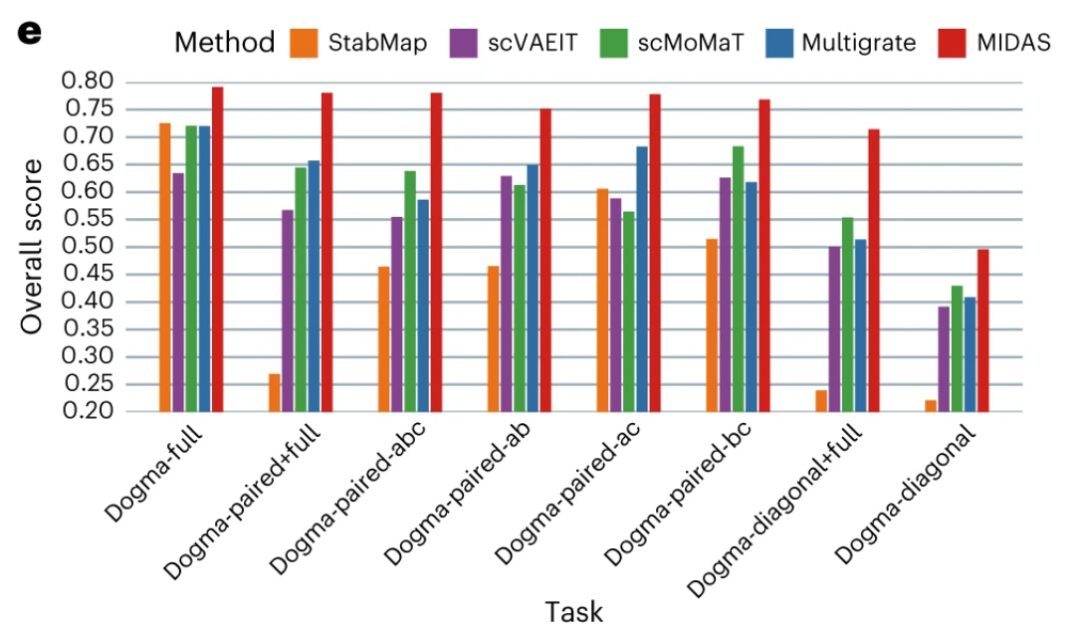
パフォーマンスの定性的および定量的な評価スコア
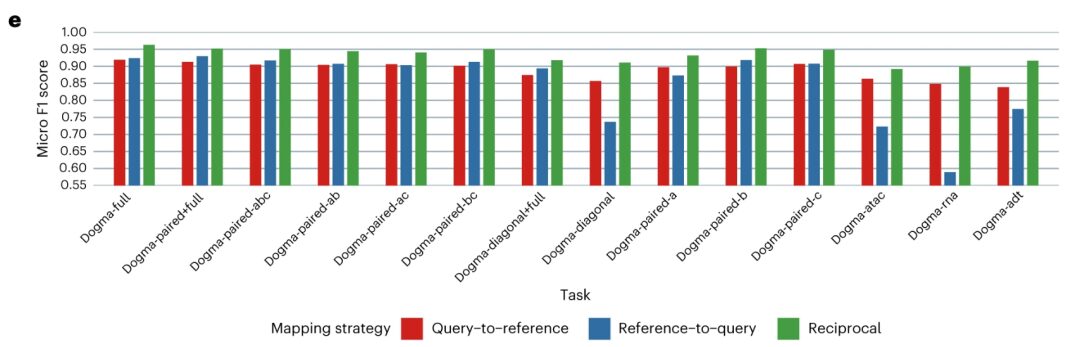
知識伝達機能の観点から、研究者は各クエリ データ セットを参照データ セットと調整し、それを k 近傍と比較します (kNN) アルゴリズムはセルタイプラベルを転送します。生物学的状態をマッピングして視覚化した後、さまざまなクエリ データ セットの相互パラメータ マッピングの結果は一貫しており、ドグマフル データ セットを通じて得られたマップ統合の結果と高度に一致していることがわかります。 MIDAS は堅牢かつ正確なラベル転送を可能にし、de novo 統合や下流分析の必要性を排除します。したがって、MIDAS を使用すると、ゼロからの高価なトレーニングや複雑な下流分析を行わずに、アトラス レベルの知識をさまざまな形式のユーザー データセットに転送できます。
要約すると、MIDAS は、単一細胞モザイク データ生成プロセスをモデル化することで、入力から生物学的状態と技術的ノイズを正確に分離し、モダリティを堅牢に調整して、マルチソースおよび異種アンサンブル分析をサポートできます。 MIDAS は、正確で堅牢な結果を提供し、さまざまなモザイク統合タスクを実行する際に他の方法よりも優れたパフォーマンスを発揮します。
さらに、MIDAS は、参照データセットからクエリ データセットに知識を効率的かつ柔軟に転送し、新しいマルチオミクス データの便利な処理を可能にします。優れた次元削減とバッチ補正パフォーマンスを備えた MIDAS は、正確な下流生物分析を可能にします。 MIDAS は、モザイク データのクラスタリングと細胞型の識別を可能にすることに加えて、連続状態の細胞の擬似時間解析も支援します。これは、RNAomics データが利用できない場合に特に価値があります。異なる組織間で知識を伝達する際、MIDAS は異種データセットを調整し、新しい細胞であっても細胞の種類を識別できます。
単一細胞マルチオミクス解析は発展を続けています
一粒の砂から世界を見ることができるのと同じように、科学者は小さな細胞の中から多宇宙、より正確には「マルチオミクス」を見ることができます。
ゲノム、トランスクリプトーム、エピゲノム、および個々の細胞のその他の特性は、さまざまな技術を使用して研究されます。各技術はそれ自体でも有益ですが、それらを組み合わせた分析 (つまり、マルチオミックス分析) により、より完全な全体像が得られます。現在、細胞生物学とトランスレーショナル研究は、単一細胞マルチオミックスによって大きく進歩していますが、データの統合と分析は依然として多くの科学者にとって課題となっています。
これに基づいて、前述のイン・シャオミン・チームとボー・シャオチェン・チームに加えて、より多くの研究チームや企業が、より効率的でシンプルなデータ処理方法を模索し続けています。
例えば、10x Genomics の Chromium 単一細胞プラットフォームなどの分析方法は拡大し続けており、人々はさまざまな組み合わせで複数の細胞特性を評価できるようになります。完全なトランスクリプトーム遺伝子発現、タンパク質発現、完全長ペアリングを含む TCR BCR シーケンス、抗原特異性、およびオープン クロマチン分析。うち セルレンジャー このソリューションは、無料で使いやすい一連の分析パイプラインを使用して Chromium 単一細胞データを分析し、生データの処理、比較の実行、遺伝子のカウントを行うことができます。さらに、Cell Ranger をクラウド分析プラットフォームと統合して、データを監視、管理、処理することができます。
別の例としては、2022年5月2日、北京大学/昌平研究所のGao Ge氏の研究グループは、「グラフリンク埋め込みによるマルチオミックス単一細胞データの統合と規制推論」と題する研究論文をNature Biotechnology誌に発表した。グラフ結合戦略に基づく深層学習手法 GLUE が提案され、数百万の単細胞マルチオミクス データの教師なしでの正確な統合と規制推論を初めて達成しました。
これらのバイオインフォマティクスツールとソフトウェアの継続的な開発は、研究者が複雑なマルチオミクスデータセットを解釈し、細胞生物学の発展を支援するのに役立ち、細胞の機能と分子制御機構を明らかにし、疾患の発生と発症を研究する上で非常に重要です。 . 最終的には「人々のためになる」ことを実現します。
参考文献:
1.https://www.chinagut.cn/articles/ss/02bc1e86e3734acebff57395d6e044a6
2.https://m.ebiotrade.com/newsf/2023-10/20231023151001602.htm
3.https://news.bioon.com/article/e49a810955a1.html
4.https://m.thepaper.cn/newsDetail_forward_26137031








