Command Palette
Search for a command to run...
中山大学の李華山氏と王彪氏の研究グループは、材料特性を高精度で予測するためのSEN機械学習モデルを開発した

内容の概要: 大域的な結晶対称性の理解と等変情報の分析は、材料特性を予測するために重要ですが、畳み込みネットワークに基づく既存のアルゴリズムでは、これらのニーズを完全に満たすことはできません。これに応えて、中山大学の李華山氏と王彪氏の研究グループは、固有の結晶対称性と材料構造クラスター間の相互作用を正確に認識する、SENと呼ばれる機械学習モデルを開発した。
キーワード: 材料性能予測ディープラーニング MP データベース
著者 | 李宝珠
編集者 | 三陽
結晶の対称性は、材料の物理的特性の研究、結晶構造の理解、新しい材料の設計、および X 線回折などの実験の実行において重要な役割を果たします。結晶の対称性を理解すると、解析が簡素化され、材料特性がより深く理解され、材料特性の計算がより効率的に行われるようになります。さらに重要なことに、結晶の対称性は、材料の電荷分布、光学的特性、磁気的特性、その他の物理的特性にも直接影響を与える可能性があります。
近年、機械学習の観点からは、結晶の対称性を材料の不変性と均等な変形とみなすことができる機械学習が広く使われていますが、現在では高度なグラフネットワークに基づいた結晶材料の機械学習が行われています。アルゴリズムは、複雑な材料の不変性と等価性をエンコードするのが困難です。
さらに、スタックド カプセル オートエンコーダー (SCAE) は元のデータから空間対称特徴を直接抽出することもできますが、従来のカプセル モデルでは依然として複雑な材料システムの構造と性能の関係を解析できません。
上記の課題に応えて、中山大学の李華山氏と王彪氏の研究グループは、SEN(対称性強化等分散ネットワーク)と呼ばれる機械学習モデルを開発した。は、対称性の高い空間群における畳み込みベースのアルゴリズムのパフォーマンスが悪いという問題を克服し、すべての空間群で高精度の材料特性予測を実現します。現在、関連する結果は「Nature Communications」に掲載されています。

関連する結果は「Nature Communications」に掲載されています
紙を入手してください:
https://www.nature.com/articles/s41467-023-40756-2
01 データセット: MP データベース内の 6,027 個の結晶材料
研究者らは、化学環境の概念とグラフィカルモデルの表現方法に基づいて結晶材料の特性を抽出し、ターゲット原子のカットオフ半径内の周囲の原子と結合による化学環境を定義し、オープンソースのPythonであるマテリアルズから結晶材料の特性を抽出しました。プロジェクトは、材料分析用のデータベースを使用して、原子の種類、原子の接続性、および各原子の周囲の結合長を抽出します。
報告されているのは、この研究でバンド ギャップと地層エネルギーを予測するために使用されたデータ セットは、マテリアルズ プロジェクト データベースからのものです。バンド ギャップと地層エネルギーのデータ セットには、それぞれ 6,027 が含まれています (8 の比率でトレーニング セット、検証セット、テスト セットに分割されています)。 :1:1) と 30,000 の素材。2 つのデータ セットは 64 個の元素で構成され、希ガス グループ、ランタニド、アクチニド、放射性元素を除く周期表の元素をカバーしています。
研究者らは、密度汎関数理論 (DFT) 計算を使用して、マテリアルズ プロジェクト データベース内の 6,027 個の結晶材料で構成されるデータ セットを予測し、予測結論に基づいて SEN モデルのパフォーマンスをテストしました。
この研究で使用された結晶対称性と化学環境データは Zenodo データベースから入手できます。
リンクにアクセス:
https://doi.org/10.5281/zenodo.8142678
02 モデルアーキテクチャ: 3 つのモジュールの統合トレーニング
以下の図に示すように、SEN モデルは、特徴抽出 (FE)、対称認識 (SP)、特性予測 (PP) モジュールを含む複雑な深層学習アーキテクチャを採用しています。
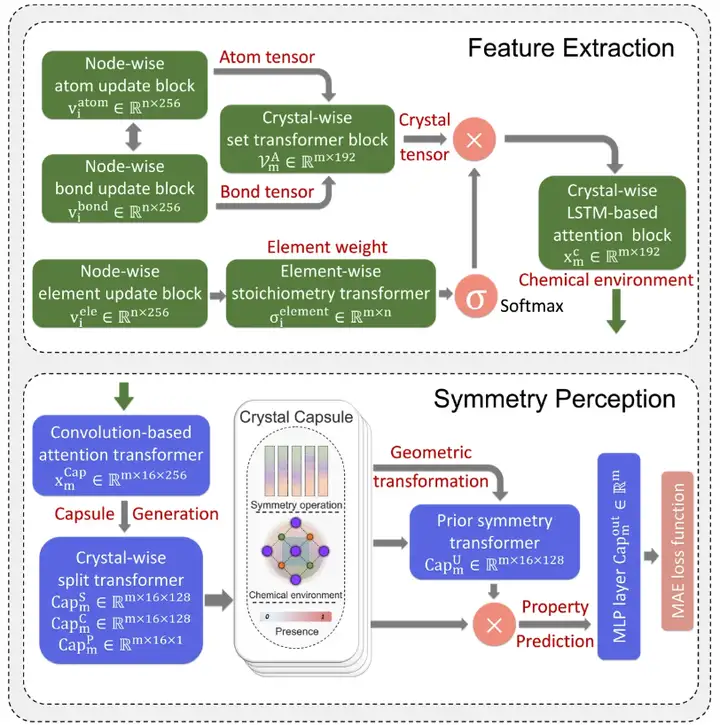
SEN アーキテクチャは、特徴抽出、対称性認識、および属性予測モジュールで構成されます。
この研究では、研究チームは3つのモジュールの統合トレーニングを通じてさまざまな材料特性の正確な予測を達成し、SENモデルを通じて原子間の相互作用を記述しました。
まず、特徴抽出モジュールが入力原子と化学結合データを感知します。入力データには、ターゲット材料の元の単位で N 個の原子と M 個の結合の情報が含まれています。最後に、ハイスループットのスクリーニングプロセスを通じて、化学量論、結晶構造、原子情報、結合情報を含む材料データセットが構築されました。
研究者らは、SEN モデルの唯一の入力データとして材料データセットを使用し、構造データと化学量論データに基づいて原子化学環境ベクトル VmA と元素重量ベクトル VmE を同時に計算しました。
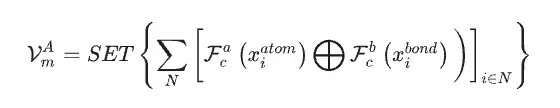
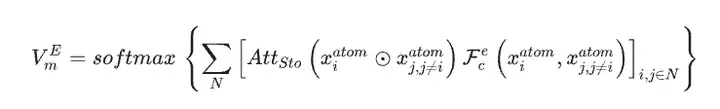
多層パーセプトロンによる活性化後、要素重みベクトルは対応するアトムの確率ベクトルに変換されます。次に研究者らは、原子化学環境ベクトルと元素重みベクトルの間の要素ごとの操作を通じてすべての原子レベルの相関関係を更新し、それによって LSTM 注目層を通じて材料の化学環境マトリックスを取得しました。
第二に、本研究は、カプセルメカニズムを材料特性予測に革新的に適用し、カプセルメカニズムに基づいて設計された対称センシングモジュールを通じて、材料の化学環境を対称演算子、畳み込み材料の化学環境と構成された材料のカプセルに変換します。結晶の対称性を感知して維持します。さらに、材料の化学環境マトリックスに対して対称操作を実行することにより、さまざまな対称パターンを結晶カプセルに一般化できます。
最後に、特性予測に関して、SEN モデルは MLP ベースのマッピング関数を通じてターゲットの材料特性を予測します。
03 SENモデルは材料特性を高精度に予測します
結論 1: SEN モデルは原子相互作用情報を正確に認識する
特徴抽出モジュールの有効性を検証するために、研究者らは、平均絶対誤差(MAE)が0.15 eV未満になるまで結晶材料のバンドギャップを予測するSENの能力を訓練し、特徴抽出モジュールによって生成された化学環境中間データを分析しました。 。
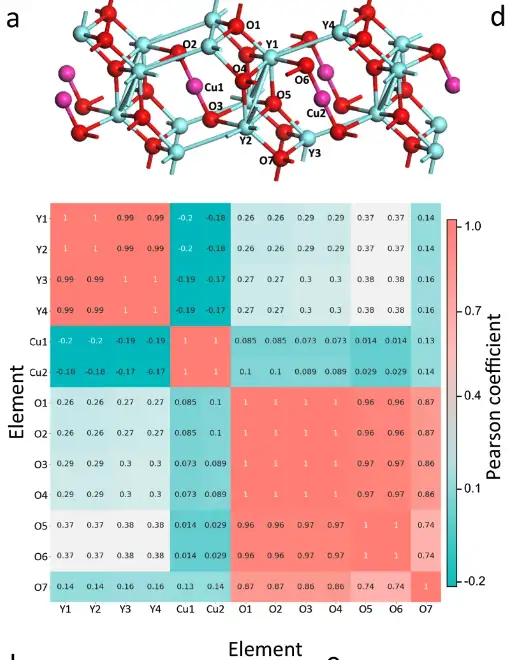
化学環境の原子ベースの相関解析
具体的には、研究者らは、Y4Cu2O7 の元のセル内の各原子の化学環境マトリックスを抽出しました。原子行列間のピアソン係数が計算され、上図に示す相関解析図が生成されました。同じ元素グループ内の原子間のピアソン係数は、異なる元素グループの原子間のピアソン係数よりもはるかに大きいため、Y4Cu2O7 の 3 つの元素グループは明確に区別できます。
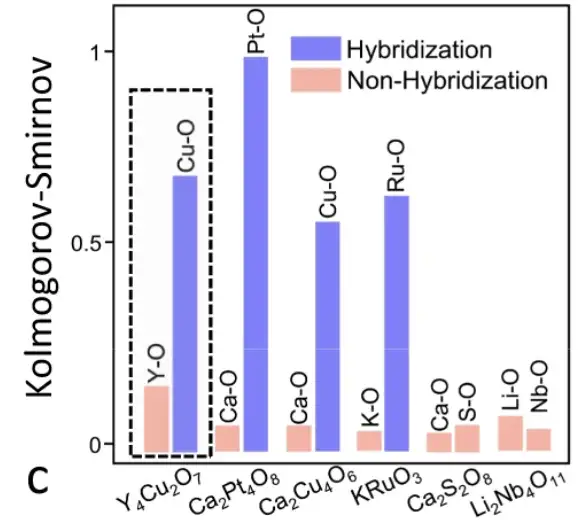
SEN モデルを通じて 6 つの材料の原子相関を学習
上図に示すように、SEN モデルは原子相互作用情報を学習およびコード化し、電子物性の予測にとって非常に重要なハイブリダイゼーション現象の検出に成功しました。
結論 2: SEN モデルの予測パフォーマンスは MegNet よりも優れています
SEN モデルにおける化学環境から材料特性へのマッピングを研究するために、研究者らは MP データベースから 5 つの材料 (Be(6)Ni(2)、Sr(4)Ge(2)S(8)、バンドギャップ) を選択しました。 Li(2)V(2)F(12)、CsAsF(6)、および BaB(2)F(8) の は、それぞれ 0 eV、3.25 eV、4.86 eV、7.24 eV、および 10.12 eV です。
バンド ギャップと材料の化学環境の PDF (確率密度関数) の間に強い相関関係があることが観察されます。つまり、バンド ギャップが増加するにつれて、PDF は徐々に拡散します。材料の化学環境からバンド ギャップまでのデータ セット全体の投影を下の図に示します。6,027 個の結晶材料が主特徴空間に均等に分布しており、バンド ギャップの変化は空間全体で連続的かつ単調です。 。
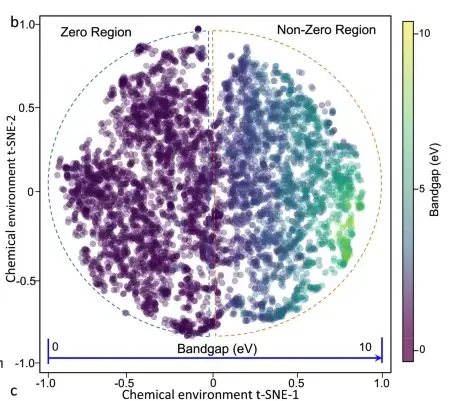
6027 材料の 2D t-SNE プロット、円の色はバンドギャップ値を示します
機械学習モデルによって学習された特徴と特性の関係が基本的な物理原理に準拠していることを検証するために、研究者らは、Ca-OX 材料の化学環境の 2D t-SNE プロットを生成し、さまざまな材料の特徴 (組成、点群、スピン極性など)、材料のバンドギャップは複雑な材料特性に依存し、1 つの重要な要素によって単純に予測できないことが最終的に発見されました。
それにもかかわらず、SEN モデルはバンドギャップ予測において大幅な改善を達成しました。テスト データ セットの材料バンド ギャップを予測する場合、SEN モデルは 0.25 eV の平均二乗誤差 (MAE) を達成しました。これは、テスト データに対して MLP、DenseNet、TFN、SE(3)、および EGNN モジュールを備えたモデルによって得られる値と同等です。 MAEと比較すると大幅な改善が見られます。
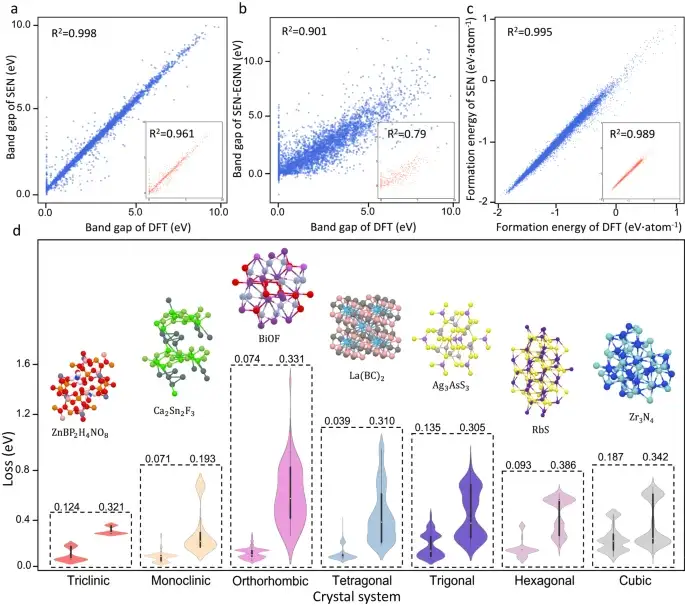
対称度の異なる結晶材料の物性予測
上の図 d に示すように、研究者らは、異なる結晶系について SEN モデルと MegNet21 モデル (一般材料ネットワーク モデル) の予測品質を比較し、材料性能予測に対する対称性認識の重大な影響をさらに明らかにしました。誤差分布プロットから、SEN モデルの予測パフォーマンスは、すべての結晶系において MegNet よりも優れています。
さらに、SEN モデルは結晶の完全な対称性を検知することにより、実効フィーチャー寸法を大幅に削減します。このフィーチャー クリーニング プロセスにより、オーバーフィッティングの問題が軽減され、材料フィーチャーからプロパティへのマッピングが強化されます。
論文はそれを示していますバンド ギャップと地層エネルギーの予測における SEN モデルの平均絶対誤差は、一般的な機械学習モデルよりそれぞれ約 22.9% と 38.3% 低くなります。
04 AIが素材産業の変革と発展を促進
長い間、新材料の設計、研究開発、材料特性の改革は科学技術の進歩を牽引する原動力の一つであり、エレクトロニクス、エネルギー、医療、航空宇宙、産業などの多くの分野で重要な役割を果たしてきました。すぐ。しかし、従来の材料の研究開発プロセスでは、継続的に性能を修正し、実現可能性を高めるために多数の実験が必要になることが多く、このプロセスには多くの人的資源と資金が必要です。
AI の応用の加速に伴い、科学のための AI はますます注目を集めており、材料との組み合わせは、ますます多くの学者や企業が模索する新しい方向性となっています。一方では、AI は大量のデータを分析してシミュレーション予測を実行できるため、新しい材料の発見と性能の最適化が加速されます。その一方で、材料科学は機械学習などの主要な AI テクノロジーの重要な基盤にもなっています。自然言語処理、ハイパフォーマンスコンピューティングのポイント。
AI は新しい材料の設計と応用を静かに変えていると言えます。今後、より強力な AI モデルが反復され、データ共有を通じて材料データベースが更新および拡張されるにつれて、AI は新たな材料の誕生をさらに促進するはずです。








