Command Palette
Search for a command to run...
SeedFold : Extension de la prédiction de la structure des biomolécules
SeedFold : Extension de la prédiction de la structure des biomolécules
Yi Zhou Chan Lu Yiming Ma Wei Qu Fei Ye Kexin Zhang Lan Wang Minrui Gui Quanquan Gu
Résumé
La prédiction de structures biomoléculaires à haute précision constitue un élément fondamental dans le développement de modèles fondamentaux biomoléculaires, et l’un des aspects les plus cruciaux de la construction de tels modèles réside dans l’identification des « recettes » permettant d’élargir l’échelle du modèle. Dans ce travail, nous présentons SeedFold, un modèle de repliement qui parvient à augmenter efficacement la capacité du modèle. Nos contributions sont triples : premièrement, nous proposons une stratégie efficace d’agrandissement de la largeur pour le Pairformer, visant à renforcer la capacité de représentation ; deuxièmement, nous introduisons un nouveau mécanisme d’attention triangulaire linéaire, qui réduit la complexité computationnelle afin de permettre un agrandissement efficace ; enfin, nous construisons un grand jeu de données pour la distillation, permettant ainsi d’élargir considérablement l’ensemble d’entraînement. Des expériences menées sur FoldBench montrent que SeedFold surpasser AlphaFold3 sur la plupart des tâches liées aux protéines.
One-sentence Summary
The authors from ByteDance Seed propose SeedFold, a scalable biomolecular structure prediction model that enhances capacity through width-scaling of the Pairformer, employs linear triangular attention to reduce computational complexity, and leverages a 26.5M-sample distillation dataset, achieving state-of-the-art performance on FoldBench and outperforming AlphaFold3 in protein-related tasks.
Key Contributions
- The paper addresses the challenge of scaling biomolecular structure prediction models by identifying width scaling of the Pairformer as a key strategy to increase representation capacity, overcoming bottlenecks posed by fixed hidden dimensions in existing architectures.
- It introduces a novel linear triangular attention mechanism that reduces computational complexity from cubic to quadratic, enabling efficient scaling without sacrificing prediction accuracy.
- The authors construct a large-scale distillation dataset of 26.5 million samples derived from AlphaFold2, significantly expanding training data and improving model generalization, with SeedFold outperforming AlphaFold3 on most protein-related tasks in FoldBench benchmarks.
Introduction
The authors leverage recent advances in large-scale model scaling to improve biomolecular structure prediction, a critical task for drug discovery and structural biology. While prior models like AlphaFold2 and AlphaFold3 achieved high accuracy, they are limited by fixed architectural choices—particularly narrow pairwise representations and computationally expensive triangular attention—that constrain model capacity and scalability. The main contribution of SeedFold is a three-pronged approach: first, it scales the Pairformer width to 512, significantly increasing representational capacity; second, it introduces linear triangular attention to reduce computational complexity from cubic to quadratic, enabling efficient scaling; third, it constructs a large-scale distillation dataset of 26.5 million samples derived from AlphaFold2 to enhance training data quality and diversity. Experiments on FoldBench show that SeedFold outperforms AlphaFold3 and other open-source models across multiple protein-related tasks, with distinct strengths in different interaction types depending on the attention variant used.
Dataset

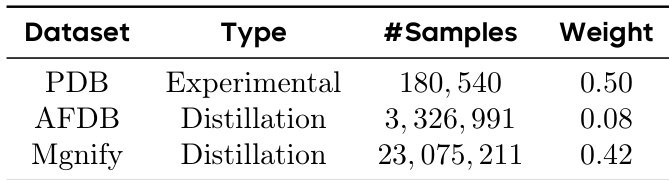
- The dataset for SeedFold consists of 26.5 million samples, expanded through large-scale data distillation from two primary sources: the experimental dataset (0.18M) and a distillation dataset derived from AFDB and MGnify.
- The experimental dataset includes PDB structures up to September 30, 2021, processed using Bolt’s pipelines, with chains and interfaces clustered; during training, samples are weighted by molecular type and cluster size to ensure balanced representation.
- The AFDB dataset contributes 3.3 million structures from AlphaFold2-predicted models on UniProt sequences. Sequences are clustered at 50% identity, and structures with pLDDT below 0.8 are filtered out. Short monomers (under 200 residues) are sampled with a probability of 0.08, followed by uniform sampling within length ranges.
- The MGnify dataset forms the core of the distillation data, comprising 23 million metagenomic sequences from environmental microbes. Sequences with fewer than 200 residues are removed, then clustered at 30% identity using MMSecs. Multiple sequence alignments (MSAs) are generated via colabfold_search using uniref30_db and colabfold_env databases. High-quality structures are inferred using OpenFold with AlphaFold2’s official weights.
- The AFDB dataset is biased toward short proteins (median length 95), while MGnify contains more diverse and longer sequences (median length 435), making it valuable for modeling long protein structures.
- During training, the model samples from the experimental and distillation datasets with equal probability. The distillation data is used in continual training stages, and removing it leads to significant drops in intra-protein lDDT, demonstrating its critical role in model performance.
- No explicit cropping is applied; instead, length-based sampling strategies are used to control input size, and metadata such as sequence length, cluster ID, and pLDDT scores are constructed during preprocessing to support training weighting and filtering.
Method
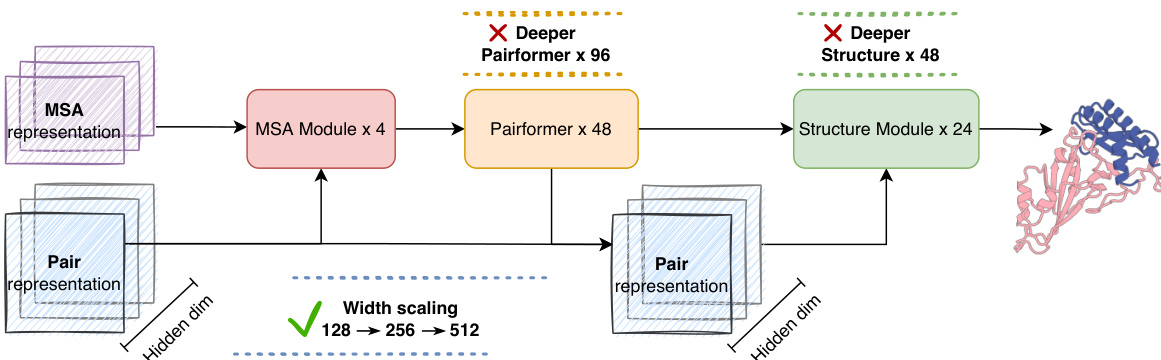
The authors leverage a modular architecture for their protein folding model, structured around a trunk and a structure module. The trunk module, responsible for encoding evolutionary and structural information, consists of an MSA module and a Pairformer module. The MSA module processes multiple sequence alignments (MSAs) across multiple recycles, extracting evolutionary features and updating the pair representation. This pair representation is then used to update the MSA hidden representation, creating a bidirectional information flow. The Pairformer module subsequently refines the pair representation through pairwise triangular multiplication and triangular attention, capturing inter-token interactions. The structure module takes the encoded pair and single representations from the trunk as conditions to perform all-atom structure generation. The overall framework is designed to explore different scaling strategies, with the model's capacity primarily bottlenecked by the hidden dimension of the pair representation. The authors investigate scaling the trunk width, trunk depth, and structure module depth to determine the most effective approach for improving performance.
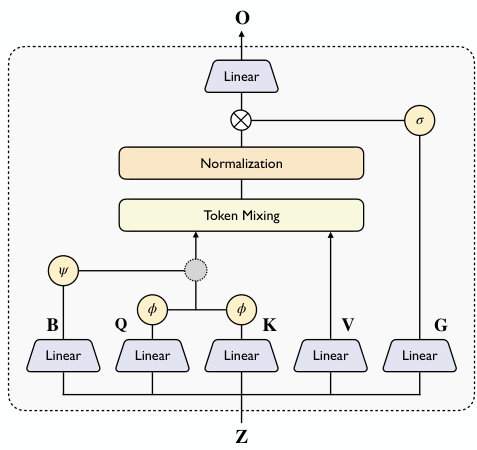
To address the computational bottleneck of triangular attention operations, the authors introduce a novel Linear Triangular Attention module as a drop-in replacement for the vanilla triangular attention. This module aims to reduce complexity by incorporating linear attention, a memory-saving technique. The standard triangular attention operation updates the pair representation Z∈Rn×n×d by computing attention scores for each row and column, which requires materializing a large n×n matrix, leading to a complexity of O(n3d). The linear attention reformulation replaces the softmax kernel with a non-linear feature map, enabling the use of the right product trick to reduce the computational cost from O(n2d) to O(nd2). However, the original linear attention formulation does not incorporate the bias term, which is essential for geometric reasoning in 3D space. The authors propose a triangular form of linear attention that reweights the attention scores by incorporating the bias term B. They present two variants: Additive Linear Triangular Attention, which uses an additive operation to up-weight or down-weight the attention score, and Gated Linear Triangular Attention, which uses a multiplication operation as a gating mechanism to control information flow. The architecture of this module is illustrated in the figure below.
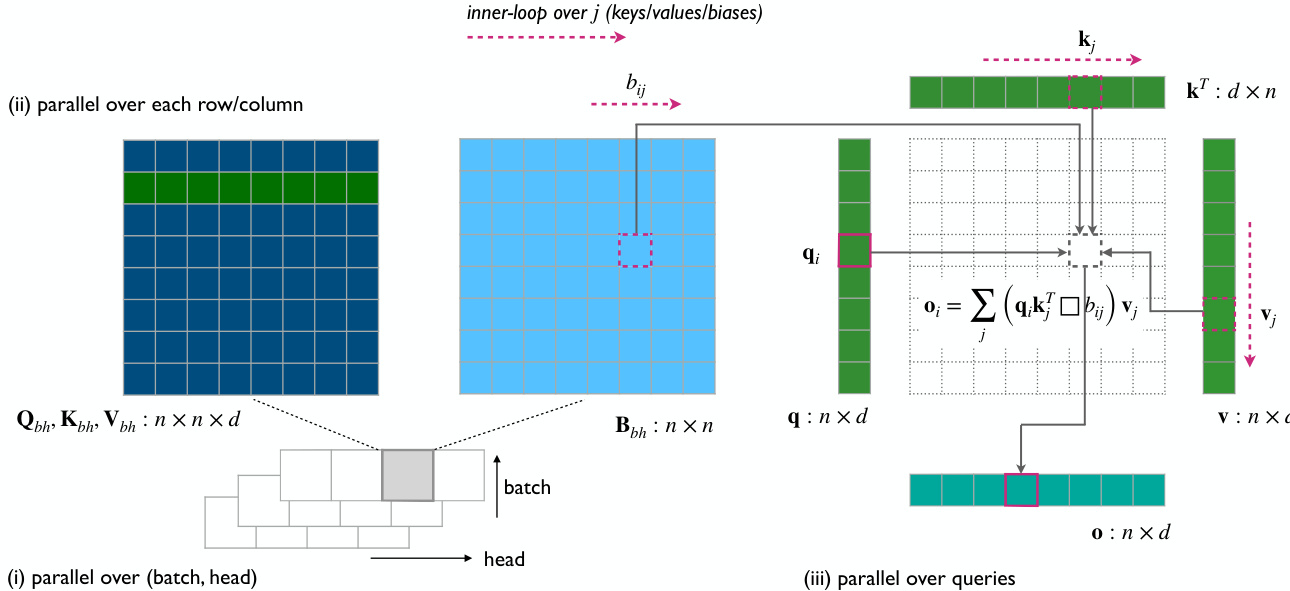
The Gated Linear Triangular Attention variant, which uses a pointwise matrix multiplication, disrupts the matrix chain product, preventing the use of the right product trick. To mitigate this, the authors develop a tiled version of linear attention optimized for reducing memory footprint on CUDA devices. The implementation of this module is further detailed in the Triton kernel, which computes the output O and its gradients. The kernel treats the batch and head dimensions as a single batch dimension and parallelizes over the first dimension of size n, iterating over the second dimension to aggregate the result and avoid materializing the full n×n matrix. The architecture of the Linear Triangular Attention module includes a normalization and gating step on the output, following the Lightning Attention design, which has proven effective in large language models. The figure below illustrates the implementation of this kernel.
Experiment
- Two-stage training strategy validates improved model performance: Stage 1 (384-token crops, 64 diffusion batch) enables fast exploration, while Stage 2 (640-token crops, 32 diffusion batch) enhances long-sequence modeling, achieving state-of-the-art results on FoldBench.
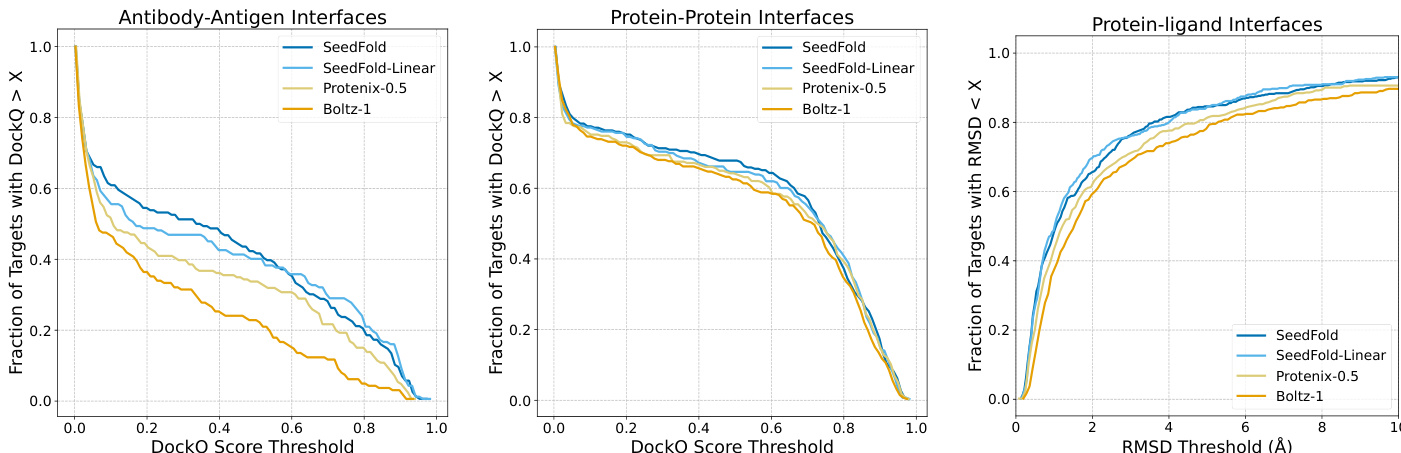
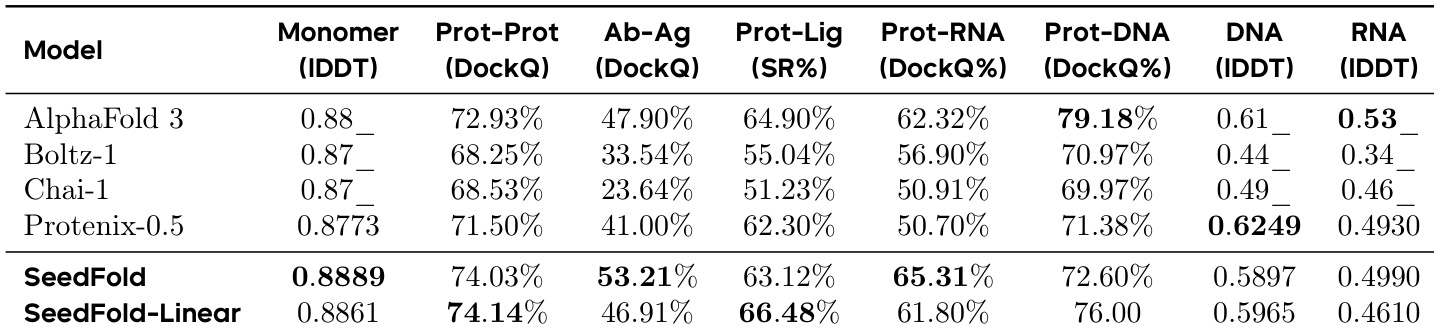
- SeedFold (512-width, vanilla triangular attention) achieves 0.8889 lDDT on monomers and 53.21% DockQ on antibody-antigen interfaces, outperforming AlphaFold 3; SeedFold-Linear (384-width, GatedLinearTriAtt) leads in protein-ligand (66.48% success) and protein-protein interfaces (74.14% DockQ).
- Ablation studies confirm GatedLinearTriAtt’s superiority in nucleic acid-related tasks (protein-RNA/DNA) and interface prediction, with consistent gains over AdditiveLinearTriAtt and vanilla attention.
- Monomer distillation data is critical: removing it causes immediate degradation in intra-protein structure accuracy, highlighting its role in maintaining model robustness.
- Training stability techniques—extended warmup (3000 steps) and reduced learning rate (0.001)—enable successful convergence of large models, with optimized Large model showing stable training dynamics and better validation performance than Large-Raw.
The authors use a two-stage training strategy and precision-aware optimization to train SeedFold models, achieving state-of-the-art performance on FoldBench. Results show that SeedFold sets new benchmarks in protein monomer prediction and antibody-antigen interfaces, while SeedFold-Linear leads in protein-ligand and protein-protein interface prediction, demonstrating the effectiveness of both width scaling and linear attention mechanisms.
The authors use a weighted sampling strategy for training data, with AFDDB and Mgnify distillation datasets contributing significantly more samples than the PDB experimental dataset. The results show that the model's performance is influenced by the relative weight of each dataset, with higher weights assigned to distillation data to emphasize learning from structured, high-quality examples.
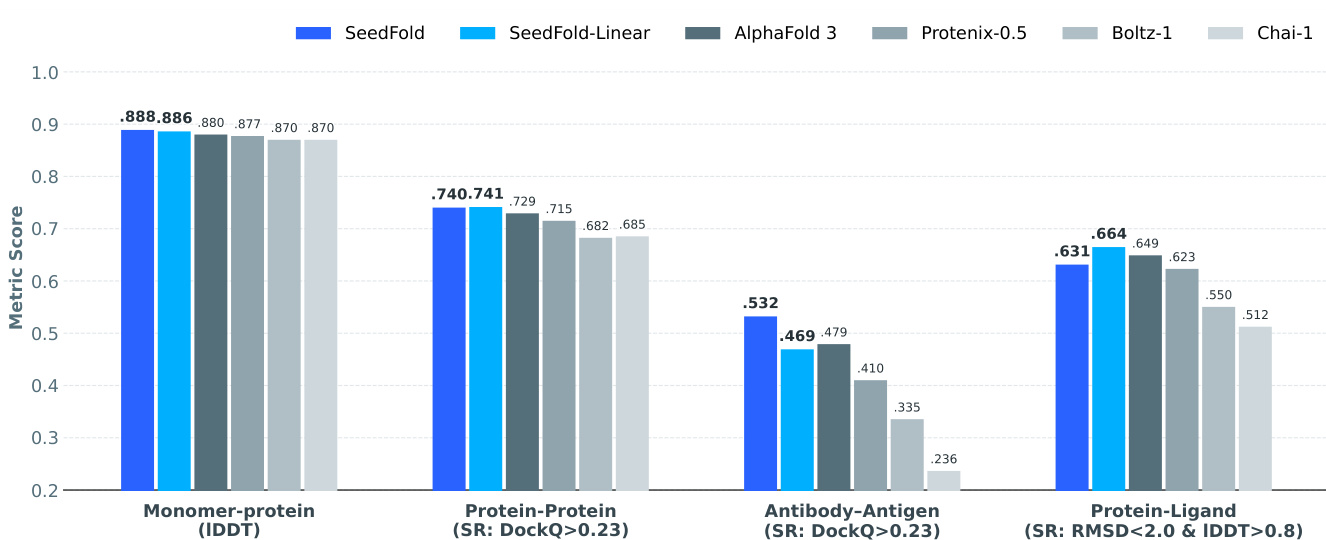
Results show that SeedFold and SeedFold-Linear achieve state-of-the-art or highly competitive performance across most FoldBench tasks, with SeedFold leading in protein monomer and antibody-antigen interface prediction, while SeedFold-Linear excels in protein-ligand and protein-protein interface prediction. The models consistently outperform AlphaFold 3 and other open-source methods, demonstrating the effectiveness of width scaling and the use of GatedLinearTriAtt for improved accuracy and efficiency.
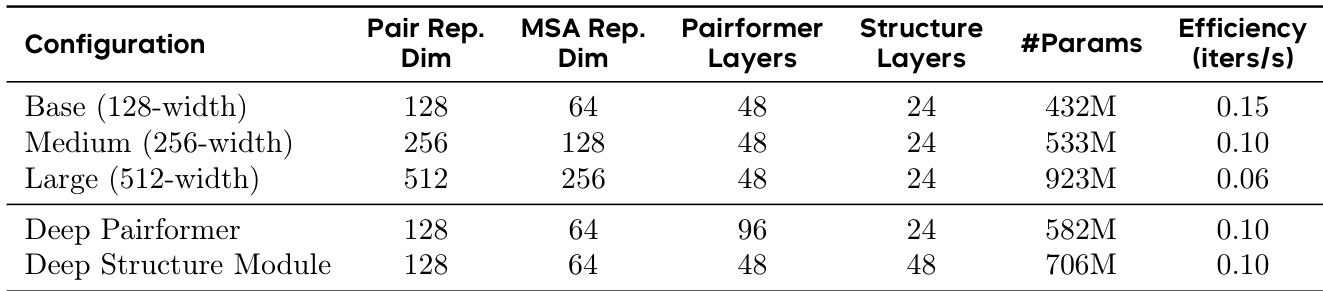
The authors use a scaling strategy to increase model capacity, showing that larger models with higher width and more parameters achieve greater performance, as seen in the transition from Base to Large configurations. Results show that increasing the Pairformer width significantly impacts efficiency, with the Large model achieving lower iterations per second compared to smaller variants, while the Deep Pairformer and Deep Structure Module configurations demonstrate improved capacity at the cost of computational speed.
Results show that SeedFold achieves superior performance in antibody-antigen interface prediction, outperforming all other models across the entire DockQ score range. In protein-protein and protein-ligand interface predictions, both SeedFold and SeedFold-Linear maintain a consistent advantage over Protenix-0.5 and Boltz-1, with SeedFold-Linear achieving the best performance in protein-ligand tasks.