Command Palette
Search for a command to run...
Sélectionné Pour l'ICLR 2025 ! Le Modèle Celcomen Proposé Par l'Université De Cambridge Permet Pour La Première Fois d'identifier l'inférence Causale Dans l'analyse Transcriptomique Spatiale
En biologie, le profil d’expression génétique d’une cellule code des informations sur ses propriétés intrinsèques et sur le microenvironnement tissulaire externe. Il est essentiel de démêler la relation de cause à effet entre ces deux effets pour comprendre pleinement les interactions complexes au sein et entre les cellules. À cette fin, un cadre solide de démêlage causal est nécessaire.
Le découplage causal est une méthode d'apprentissage automatique qui vise à séparer les fonctionnalités utiles des fonctionnalités non pertinentes en révélant des relations causales dans les données, réduisant ainsi la dépendance du modèle aux corrélations parasites et améliorant la robustesse et la capacité de généralisation du modèle. Parallèlement au développement de théories d’apprentissage automatique telles que le découplage causal, les avancées technologiques dans le domaine de la biologie ont également favorisé le développement de la transcriptomique spatiale, permettant aux chercheurs de mesurer simultanément l’expression génétique et les coordonnées spatiales des cellules à une résolution unicellulaire, et de réaliser des expériences de perturbation telles que l’inactivation de gènes à grande échelle dans des échantillons spatiaux.
Cependant,Les approches informatiques actuelles de la transcriptomique spatiale négligent souvent la modélisation des perturbations causales aux niveaux cellulaire et tissulaire.Cela est crucial pour découvrir les mécanismes à l’origine des états pathologiques dans les tissus. Par exemple, le modèle de cellules virtuelles peut prédire les effets des changements dans le microenvironnement et le macroenvironnement (tels que l'âge du donneur, le tissu cellulaire, le traitement médicamenteux, l'inactivation de gènes médiée par l'ARNg, etc.) sur l'expression des gènes, et le modèle de tissus virtuels peut non seulement estimer l'impact de l'environnement sur une seule cellule, mais également déduire l'impact d'une seule cellule sur son environnement environnant et sur l'ensemble du tissu.
Sur cette base,Une équipe de recherche de l'Université de Cambridge a proposé un modèle de tissu virtuel appelé Celcomen, qui est essentiellement un nouveau réseau neuronal graphique basé sur la causalité mathématique pour percer les secrets de la régulation des gènes intracellulaires et intercellulaires dans la transcriptomique spatiale et les données unicellulaires.Les chercheurs ont validé la capacité de Celcomen à démêler et à récupérer les interactions gène-gène dans les données transcriptomiques spatiales réelles et auto-simulées.
Les résultats associés ont été sélectionnés pour l'ICLR 2025 sous le titre « Estimation de l'effet de perturbation des cellules uniques et des tissus dans la transcriptomique spatiale via le désenchevêtrement causal spatial ».
Points saillants de la recherche :
* L'étude prouve la faisabilité d'étendre le modèle de cellule virtuelle au modèle de tissu virtuel
* L'étude propose le premier modèle causalement identifiable dans l'analyse transcriptomique spatiale
* Déduire la régulation des gènes en intégrant des données unicellulaires dissociées et des données unicellulaires spatiales
Adresse du document :
https://openreview.net/forum?id=Tqdsruwyac
Le projet open source « awesome-ai4s » rassemble plus de 200 interprétations d'articles AI4S et fournit des ensembles de données et des outils massifs :
https://github.com/hyperai/awesome-ai4s
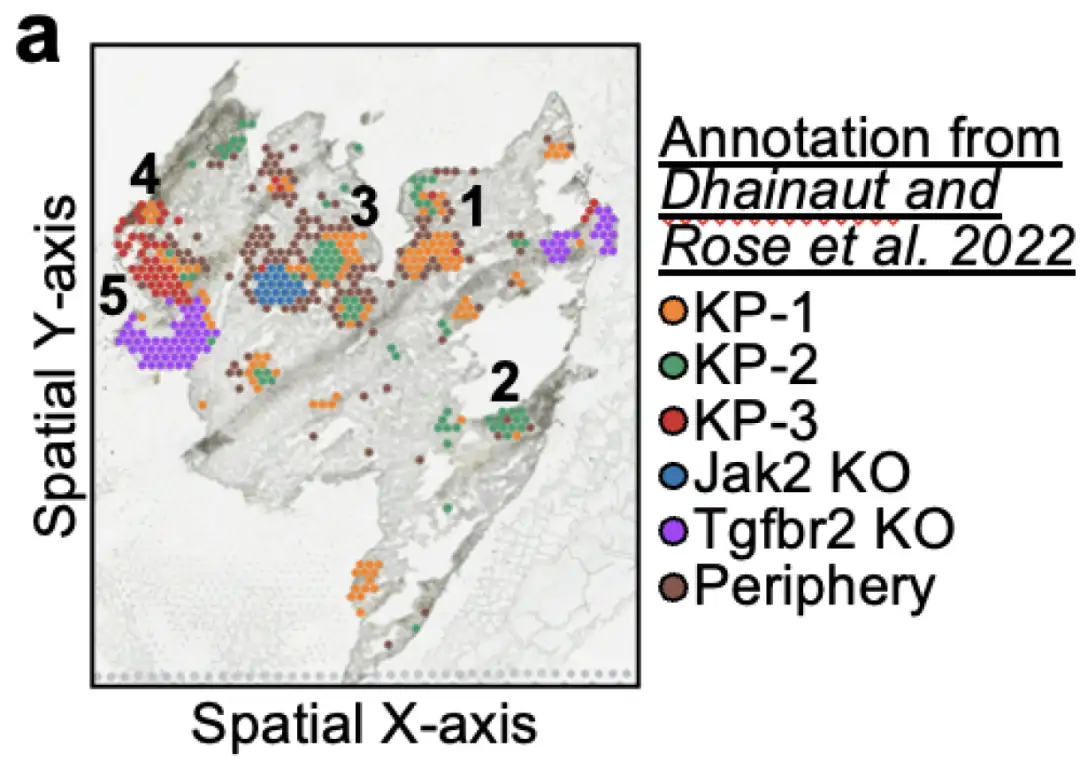
Ensemble de données : Première tentative d'utilisation de l'ensemble de données Perturbmap
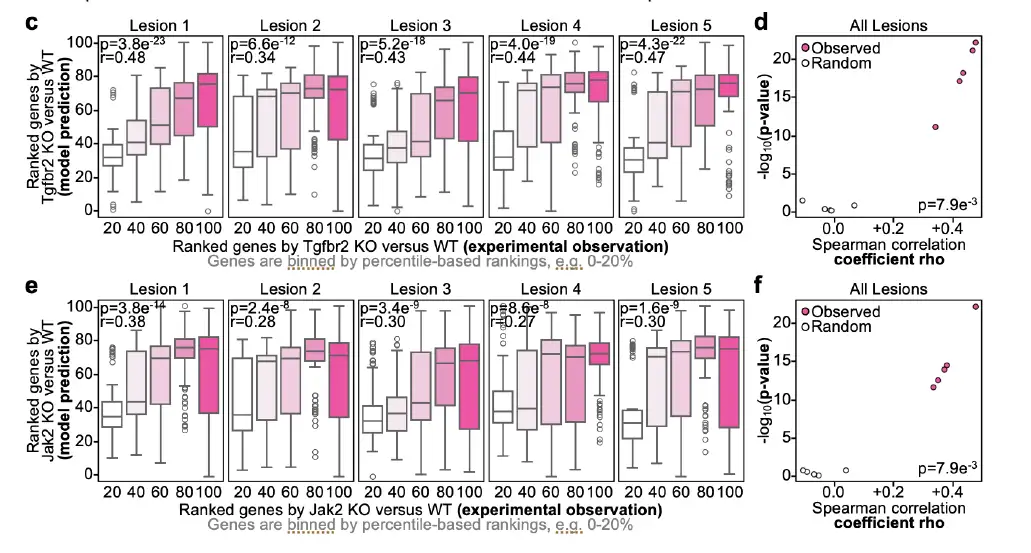
Pour démontrer l’efficacité de Celcomen dans la capture correcte des effets de perturbation dans un contexte spatial, les chercheurs l’ont comparé à un ensemble de données de transcriptome entier in vivo qui mesure la suppression des gènes dans la transcriptomique spatiale.Cela s'appelle Perturbmap. L'ensemble de données Perturbmap contient un modèle de souris pour étudier le cancer du poumon KP et, en outre, d'éventuels knockouts de Jak2 ou Tgfbr2. L'ensemble de données annote 5 régions spatiales comme régions de lésion, qui font partie de 1) cancer de type sauvage KP, ou 2) cancer KP avec knockout Jak2, ou 3) cancer KP avec knockout Tgfbr2, comme indiqué ci-dessous :
Dans le cadre de l'évaluation des capacités de Celcomen,L'ensemble de données sur la rate fœtale utilisé par les chercheurs provient de https://developmental.cellatlas.io/fetalimmune,Fourni sous forme log-normalisée, il est clair que la transformation logarithmique et la normalisation de la taille de la bibliothèque ont été effectuées ;Ensemble de données sur le glioblastome de 10x Genomics,La même normalisation de la taille de la bibliothèque, les mêmes comptages par million (CPM) et la même transformation logarithmique en base e ont été effectués ; de plus, seuls les gènes exprimés dans au moins 100 cellules ont été conservés.
Architecture du modèle : un nouveau cadre d'analyse causale Celcomen
Le modèle de Celcomen proposé dans cette étude permet d'identifier l'inférence causale et d'obtenir une meilleure interprétabilité du modèle en combinant la mécanique lagrangienne et l'inférence causale. En termes simples, l'identifiabilité signifie que le modèle peut clairement identifier les relations causales à partir de données suffisantes et d'hypothèses raisonnables, plutôt que de conduire aux mêmes résultats d'observation en raison de multiples hypothèses ou paramètres de modèle différents - cela fournit un nouveau cadre d'analyse causale pour la recherche en transcriptomique spatiale.
Celcomen repose sur trois hypothèses fondamentales : 1. La corrélation gène-gène attendue entre les voisins de premier ordre doit correspondre exactement aux données observées ; 2. La corrélation gène-gène attendue au sein du même point spatial/cellule doit correspondre exactement aux données observées ; ③ Hypothèse de suffisance causale : Il n’existe aucune cause commune non mesurée entre les paires de gènes étudiées.
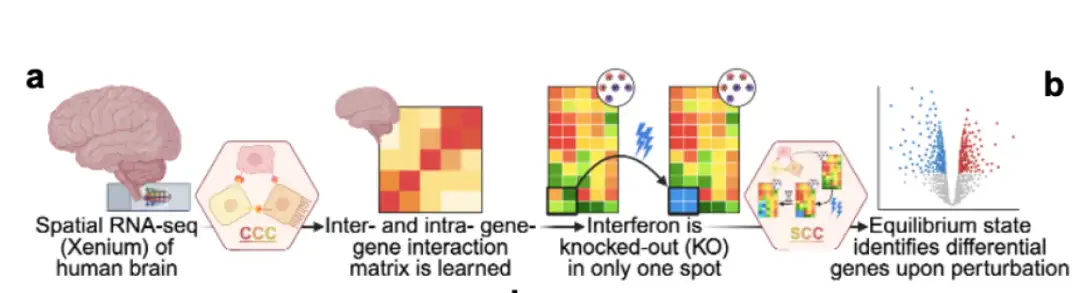
Comme le montre la figure suivante :Celcomen est divisé en deux parties : le module de raisonnement (CCE) et le module de génération (SCE) :
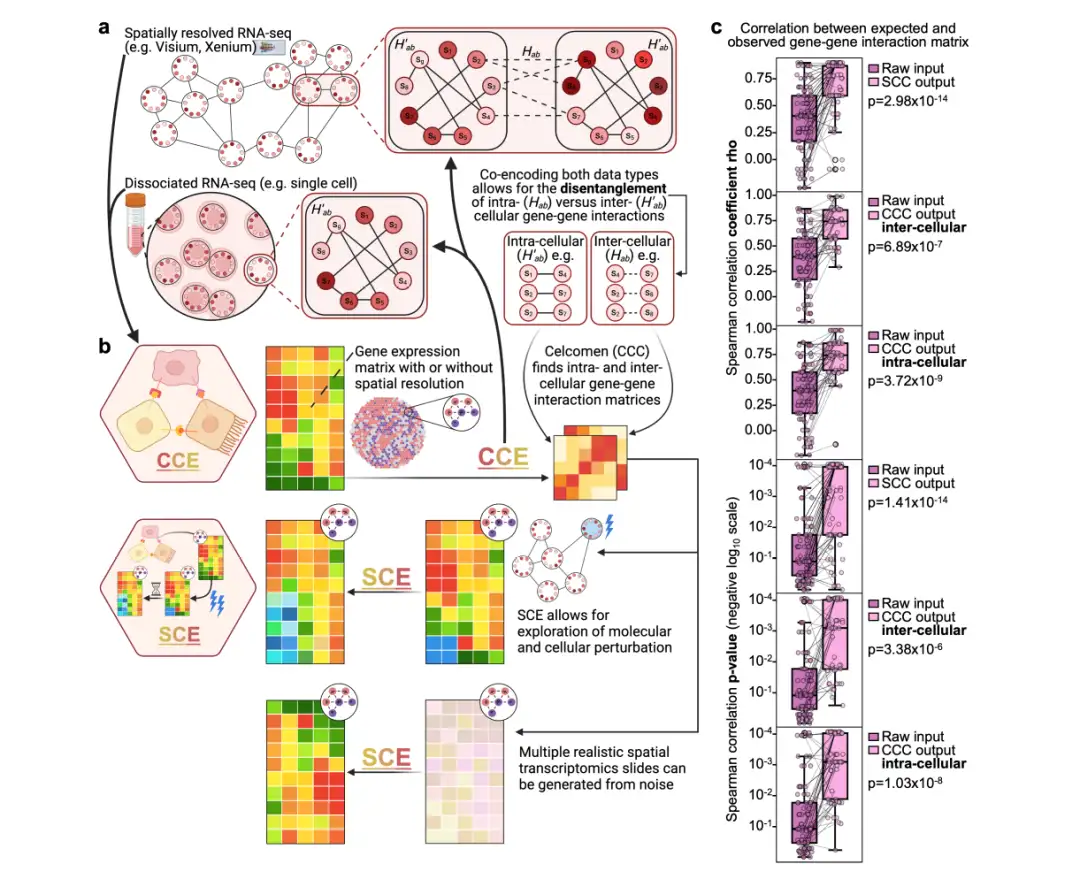
(a) Module de raisonnement (CCE) :Les relations gène-gène peuvent être apprises à partir de données de transcriptome résolues spatialement (données RNA-seq spatiales) et éventuellement à partir de données RNA-seq unicellulaires dissociées (données scRNA-seq dissociées). Les paires cellule-cellule mises en évidence dans les données spatiales et les cellules individuelles dans les données RNA-seq à cellule unique démontrent comment le CCE peut distinguer les interactions gène-gène intracellulaires (H′ab) des interactions intercellulaires (Hab).
(b) Module de génération (SCE) :Exploitez les relations gène-gène apprises par CCE pour simuler un comportement tissulaire contrefactuel après des perturbations cellulaires ou génétiques.
* Scénarios contrefactuels : Il s'agit d'une méthode utilisée pour étudier le comportement possible des tissus biologiques dans différentes conditions hypothétiques, principalement utilisée dans l'inférence causale, la simulation d'intervention et la modélisation biomédicale. Il s’agit de construire un scénario hypothétique sur la manière dont le comportement d’un organisme biologique pourrait différer de ce qui est réellement observé si un facteur clé était modifié (par exemple, suppression d’un gène, intervention médicamenteuse, changement de l’environnement externe, etc.).
Résultats de la recherche : le modèle de Celcomen est identifiable dans la démêlage des relations causales
Les chercheurs ont vérifié l’identifiabilité du modèle Celcomen dans l’apprentissage des structures causales et le démêlage des relations causales au moyen d’expériences sur des données synthétiques auto-cohérentes et des données du monde réel.
Celcomen a une forte cohérence et une forte identifiabilité
Comme le montre la figure ci-dessous, sur l'ensemble de données synthétiques, Celcomen montre systématiquement une forte cohérence entre ses interactions gène-gène déduites et les données réelles, indiquant que Celcomen a une forte auto-cohérence et donc une identifiabilité.
* Auto-cohérence : dans les statistiques, l'optimisation et l'apprentissage automatique, l'auto-cohérence signifie généralement que les hypothèses, les dérivations et les processus d'optimisation du modèle peuvent converger vers une solution stable.
* Identifiabilité : fait référence à la question de savoir si les paramètres du modèle ou les effets causaux de la relation causale peuvent être déterminés de manière unique sur la base des données observées dans le modèle d'inférence causale.
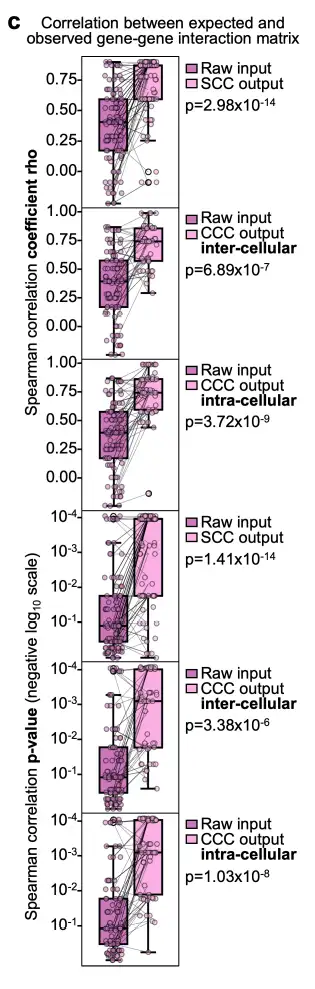
Les chercheurs ont également confirmé les garanties d'identifiabilité du modèle Celcomen sur des données humaines réelles en l'appliquant à des sections de transcriptome spatial de plusieurs rates fœtales humaines, et ont observé des coefficients de corrélation de Spearman entre les deux matrices d'interaction gène-gène dans la plage de 0,5 à 0,6. De plus, les interactions génétiques capturées sont biologiquement plausibles dans les matrices intra- et intercellulaires car elles suivent des processus biologiques intra- et intercellulaires connus.
Cela démontre l’identifiabilité de Celcomen, confirmant sa stabilité et sa robustesse implicites au-delà des données théoriques et synthétiques et peut également être observé dans des échantillons humains réels.
Capacité de découplage causal : Celcomen peut démêler avec succès les sources de variation intrinsèque et extrinsèque du transcriptome
Les chercheurs ont ensuite testé la capacité de Celcomen à démêler les programmes de régulation des gènes au sein et entre les cellules (capacité de découplage). Ils ont appliqué Celcomen dans un contexte clinique humain réel pour analyser un ensemble de données de transcriptome spatial à résolution unicellulaire du glioblastome humain (cancer du cerveau), comme le montre la figure ci-dessous. Les chercheurs ont découvert que Celcomen était capable de démêler avec succès les sources de variation intrinsèque et extrinsèque du transcriptome.
Validation contrefactuelle spatiale in vivo : Celcomen obtient des résultats significativement meilleurs qu'une base de référence aléatoire
Pour démontrer davantage l’efficacité de Celcomen, les chercheurs ont mené un test de référence sur l’ensemble de données du transcriptome in vivo Perturbmap. Les résultats ont montré que pour toutes les lésions, la corrélation de Spearman entre les prédictions et les mesures in vivo variait de 0,28 à 0,47. Pour évaluer l’importance de cette performance, les chercheurs ont comparé le modèle à une base de référence aléatoire, où Celcomen a été exécuté sur des données mélangées de manière aléatoire. Les résultats montrent que Celcomen est significativement plus performant que la ligne de base aléatoire avec une valeur p de 0,0079, comme le montre la figure ci-dessous (cf) :
En résumé, le modèle proposé dans cette étude ouvre une nouvelle voie pour parvenir à une explicabilité mécaniste par l’inférence causale. Comme démontré dans les expériences, grâce à l'identifiabilité causale du modèle Celcomen, les chercheurs peuvent récupérer les valeurs des paramètres du réseau neuronal avec une grande précision. Les avancées de Celcomen ont des impacts significatifs sur le domaine biomédical, par exemple en révélant comment la maladie provoque une défaillance tissulaire et en facilitant des hypothèses testables sur le bénéfice des traitements. La valeur de Celcomen continuera de croître à mesure que la technologie progressera, entraînant des améliorations dans la modélisation des maladies et la compréhension mécaniste.
L'intelligence artificielle libère le potentiel de la transcriptomique spatiale
Les résultats pertinents obtenus dans cette étude constituent un autre développement de la transcriptomique spatiale - la technologie de la transcriptomique spatiale est l'une des avancées majeures dans le domaine de la bioinformatique ces dernières années. Cette technologie a considérablement changé le paradigme de la recherche biomédicale en fournissant des caractéristiques moléculaires détaillées et localisées spatialement, permettant aux chercheurs en biologie d’élucider la structure et la fonction des tissus avec une résolution sans précédent.
Au cours des dernières années, la technologie de transcriptomique spatiale a connu un développement rapide et les données ont été continuellement accumulées. Sur cette base, l'article « Nature Methods Special Issue Comment: Using the "Key" of Artificial Intelligence to Open the "Lock" of Spatial Omics » publié en août 2024 soulignait queL’intelligence artificielle a le potentiel de libérer tout le potentiel de l’omique spatiale, facilitant l’intégration d’ensembles de données complexes et la découverte de nouvelles perspectives biomédicales.
Plus précisément, l’IA peut faciliter l’intégration de la transcriptomique spatiale et du scRNA-seq, permettant aux chercheurs de mesurer les profils d’expression génique spatiale à l’échelle du transcriptome au niveau de la cellule unique. De plus, en intégrant les données d’imagerie omique spatiale et histologique, l’IA peut construire des cartes tissulaires spatiales tridimensionnelles complètes et à haute résolution couvrant un large éventail de modalités omiques. À mesure que le nombre d'ensembles de données disponibles augmente, les modèles multimodaux de grand langage (MM-LLM) peuvent être formés sur des données omiques spatiales, d'imagerie médicale et de texte clinique pour des tâches de recherche biomédicale et de médecine de précision.
Octobre 2023Le groupe de recherche de Zhang Shihua à l'Institut de mathématiques et de sciences des systèmes de l'Académie chinoise des sciences, a publié un article dans Nature Computational Science.Publication d'un article de recherche intitulé « Intégration des données transcriptomiques spatiales dans différentes conditions, technologies et stades de développement ». Ce travail a établi un nouvel outil d'analyse intégré, STAligner, pour les données de transcriptome spatial de plusieurs tranches de tissus biologiques provenant de différentes technologies, de différents moments de développement et de différentes pathologies. Cela peut aider les chercheurs à découvrir de nouvelles informations biologiques importantes lors de la réalisation d’analyses transcriptomiques spatiales.
*Article original :
https://www.biorxiv.org/content/10.1101/2022.12.26.521888v1.full.pdf
Afin de résoudre les défis multiformes auxquels est confrontée l'analyse des données du transcriptome spatial, en juillet 2024,Groupe de recherche du professeur associé Zhang Qiangfeng à l'École des sciences de la vie, Université Tsinghua/Centre d'innovation avancée en biologie structurale/Centre conjoint des sciences de la vie Tsinghua-Université de Pékin,Un article de recherche intitulé « Découverte de modules tissulaires dans les données de transcriptomique spatiale à résolution unicellulaire via l'intégration cellulaire consciente de l'interaction cellule-cellule » a été publié en ligne dans la revue Cell Systems. Cette étude a développé un algorithme d'intelligence artificielle SPACE (analyse de données transcriptomiques spatiales via l'intégration de cellules « conscientes des interactions ») basé sur le cadre d'apprentissage profond de l'autoencodeur graphique, qui peut identifier les types de cellules spatiales et découvrir des modules tissulaires à partir de données de transcriptome spatial avec une résolution de cellule unique, et peut être utilisé pour la recherche de transcriptome spatial à grande échelle.
À l’avenir, en exploitant la puissante puissance de calcul et les algorithmes d’apprentissage profond de l’IA, les chercheurs devraient débloquer une nouvelle dimension de la transcriptomique spatiale, améliorer considérablement l’efficacité de la recherche sur les maladies, du développement de médicaments et de la médecine personnalisée, et permettre aux scientifiques d’explorer l’hétérogénéité spatiale des systèmes biologiques avec une précision sans précédent, apportant ainsi des découvertes scientifiques révolutionnaires.
Références :
1.https://openreview.net/forum?id=Tqdsruwyac
2.https://www.thepaper.cn/newsDetail_forward_28521641
3.https://www.cas.cn/syky/202310/t20231020_4981872.shtml








