Command Palette
Search for a command to run...
From "blind Screening" to "precise Positioning", the AlphaPPIMI Framework Significantly Improves Generalization Capabilities, and the Prediction Performance of PPIs Interface Regulators Surpasses Existing methods.

Within the complex regulatory networks of life, protein-protein interactions (PPIs) coordinate intracellular signaling, energy metabolism, and gene activity, fundamental to maintaining normal life. PPIs play a central role in maintaining both physiological homeostasis in health and the abnormal changes that occur during disease. Studies have shown that malfunctioning PPIs is closely associated with cancer, neurodegenerative diseases, and various infectious diseases. Therefore, the development of drugs targeting PPIs has become a key area of new drug research and development.
Early scientists studied protein-protein interactions such as MDM2-p53 and confirmed that intervening in such interactions has the potential to treat diseases, especially providing new ideas for disease targets that were previously difficult to target.However, the particularity of PPIs is that their interaction interfaces are usually relatively flat and lack clear structural features suitable for the embedding of small molecule drugs, which poses a huge challenge to drug development.Especially for newly discovered PPIs or those with limited structural information, it is more difficult to find molecules that can regulate their functions.
The researchers found that although the interface of PPIs is broad and flat, there are still some key areas - called "hot spots". They act like "switches" in the interaction and become ideal targets for drug design.
With the rapid development of artificial intelligence technologies, particularly machine learning and deep learning, the drug development process for PPIs has significantly accelerated. A variety of innovative algorithms and tools have emerged, including 2P2IHUNTER, which efficiently identifies potential PPI inhibitors; PPIMpred, which enables large-scale virtual screening; and SMMPPI, which not only predicts regulatory molecules but also demonstrates practical value in anti-COVID-19 research. Despite significant progress, challenges remain. Traditional computational methods, which rely heavily on similarity screening, struggle to fully capture the complex interaction characteristics of PPI interfaces. Furthermore, existing models have limited generalization capabilities across different protein types, hindering the efficiency of drug development for novel targets.
In recent years, Transformer-based pre-trained language models have provided new ideas for the above problems. These models can automatically learn key features from a large number of protein sequences, thereby predicting interactions more intelligently.
Based on this direction, the joint research team of China University of Petroleum and Yonsei University integrated multiple advanced technologies to build a new framework called AlphaPPIMI.This tool combines large-scale pre-trained models and adaptive learning mechanisms to address the core challenge of "discovering regulators that specifically target the PPIs interface."By fully leveraging the advantages of pre-trained large-scale models and effectively modeling complex binding patterns through a dedicated cross-attention module, the model's generalization ability across different PPIs families has been significantly improved, providing strong support for the future development of PPIs targeted drugs.
The relevant research results were published in the Journal of Cheminformatics under the title "Alphappimi: a comprehensive deep learning framework for predicting PPI-modulator interactions."

Paper address:
https://jcheminf.biomedcentral.com/articles/10.1186/s13321-025-01077-2
Follow the official account and reply "AlphaPPIMI" to get the full PDF
More AI frontier papers:
https://hyper.ai/papers
Dataset: Construction of PPIs dataset system with DLiP as the core
The study used the DLiP dataset as the training core, which contains 120 PPIs and their corresponding 12,605 unique regulators.It can also provide the sequence, three-dimensional structure and experimental activity data of each pair of protein complexes, providing all-round support for model building.
To conduct independent validation, the research team constructed two benchmark test sets from the DiPPI and iPPIDB databases. Both sets contain experimentally validated interface modulators, along with their structures and binding information. When compiling the data, the team implemented three quality controls: retaining only heterodimeric PPIs, removing samples with unclear binding sites, and limiting the scope to human PPIs. Furthermore, compounds that act on multiple targets were separated to ensure accurate annotation.
The final details of the two benchmark sets are as follows:DiPPI contains 201 regulators corresponding to 1,316 PPIs targets.Each sample has a molecular structure, protein sequence, interface structure and active tag; iPPIDB covers 2,203 regulators and 34 PPIs, and all protein sequences are from the UniProt database, ensuring data consistency.
After analyzing the physical and chemical properties of the two benchmark sets, it was found that they differed significantly in interface targeting characteristics and chemical space distribution, which would increase the difficulty of model generalization.By calculating the ECFP4 molecular fingerprint, we also found that the average Tanimoto similarity of the compounds in the two benchmark sets is very low.This indicates that the structural diversity of these compounds is relatively high.
For a certain family of PPIs, the study also selected regulators that are selective for other PPIs families as potential inactive samples, while excluding molecules with similar structures to known active regulators to reduce the risk of false negatives. Taking into account the imbalance in the number of positive and negative samples, the team downsampled the negative samples to construct a data set with a balanced number. Subsequent sensitivity analysis showed that no matter how the ratio of positive and negative samples was adjusted, the model performance was very stable and not highly dependent on the ratio. It should be noted that although there are verified inactive compounds, they were not included in the negative sample set because they are unevenly distributed and may cause data bias after inclusion.
In order to verify the practical application value of this method,The research team also screened the "PPIs-specific library" in the ChemDiv database - this library contains 205,497 compounds specifically designed for the interfacial properties of PPIs.This large-scale virtual screening demonstrated the practicality of this method in drug development scenarios.
AlphaPPIMI framework: multi-source feature extraction, bidirectional cross attention, and CDAN generalization optimization
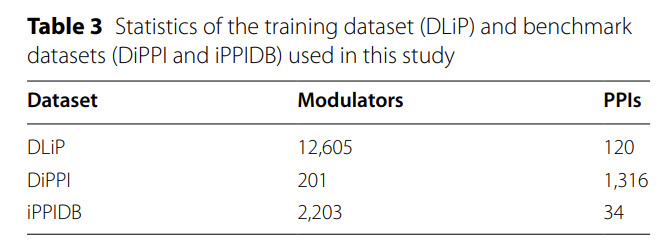
As shown in the figure below,This study developed a new computational framework, AlphaPPIMI, specifically for predicting the binding relationship between PPIs and regulators.With a particular focus on targeting interactions at interface binding sites, this framework integrates multiple advanced modules, including Uni-Mol2, ESM2, ProTrans, ECFP, and PFeature, striving to comprehensively extract PPI-related features while achieving efficient representation learning.
In the molecular characterization phase, the research team used the Uni-Mol2 model with 84 million parameters to integrate atoms, chemical bonds, geometric information and molecular fingerprints.A 768-dimensional global feature vector was generated for each modulator. The team also incorporated ECFP4 fingerprints to generate a 1,024-dimensional binary vector to capture key chemical information, such as cyclic substructure. Ultimately, these two types of features were combined to produce a 1,792-dimensional feature vector encompassing molecular topology, 3D geometry, and chemical substructure, providing reliable data support for interface binding predictions.
Protein feature extraction uses three complementary approaches:The ESM2-150M model, based on the Transformer architecture, was trained on 60 million UniRef50 sequences and generates 640-dimensional feature vectors specifically capturing amino acid relationships related to interface formation. The ProtTrans model, trained on over 45 million protein sequences, outputs 1,024-dimensional embedding vectors, capturing evolutionary patterns that complement ESM2. Finally, the PFeature method provides information on protein structure and physicochemical properties through 19 categories of descriptors. The fusion of these three methods generates a 3,366-dimensional protein representation that comprehensively covers protein sequence patterns and interface-specific properties.
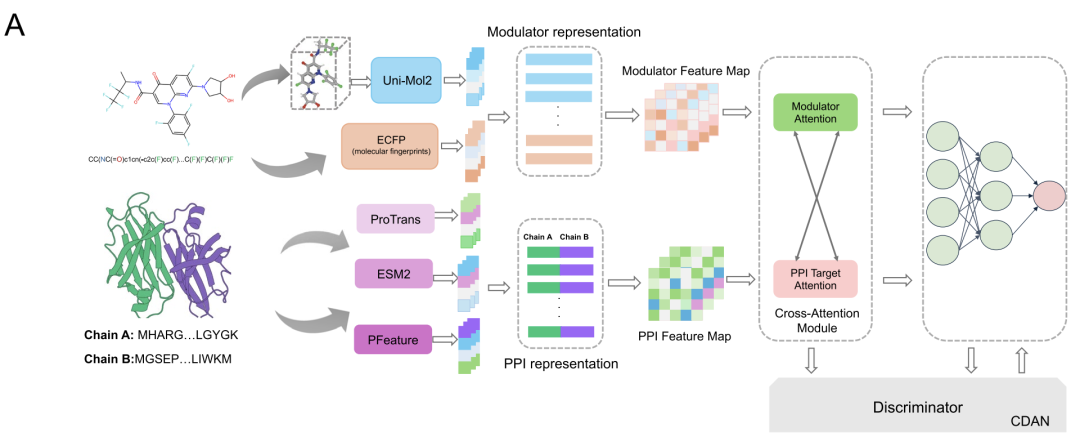
To model the complex interactions between proteins and regulators, AlphaPPIMI designed a bidirectional Cross-Attention Module, as shown in the figure below. This module first performs linear transformations on the regulator feature matrix FM and the target feature matrix FP, which are then fed into the attention submodule, enabling bidirectional information exchange at the key-value level. PPI features are optimized using the attention weights of the regulator source, while regulator features are adjusted using the PPI-driven attention mechanism.Residual connections and maximum pooling operations are also added to the module.It can dynamically learn the interaction pattern between the two while retaining the unique information of each modality, and ultimately output a more comprehensive representation of the interaction.
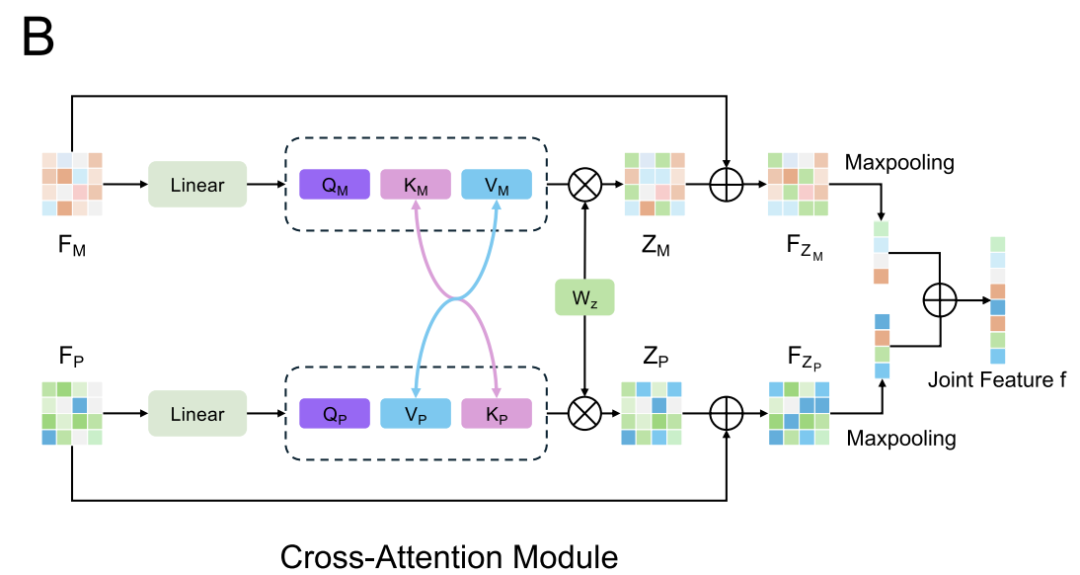
Considering the differences in feature distributions across datasets—for example, DiPPI focuses on interface-targeted modulators, while general datasets like DLiP lack such information—AlphaPPIMI also introduces a Conditional Domain Adversarial Network (CDAN), as shown in the figure below. CDAN uses a "joint representation of feature embedding and classification prediction" as a condition for the domain discriminator, preserving discriminative features while achieving distribution alignment between the source and target domains.The training process adopts a minimax game: the feature encoder and the cross-attention module are responsible for generating domain-invariant representations, while the discriminator is used to distinguish the sources of features.This mechanism significantly improves the generalization ability of the model across different protein families and provides more robust support for identifying novel interface-targeted regulators.
Evaluation and application verification of AlphaPPIMI's cross-domain generalization ability
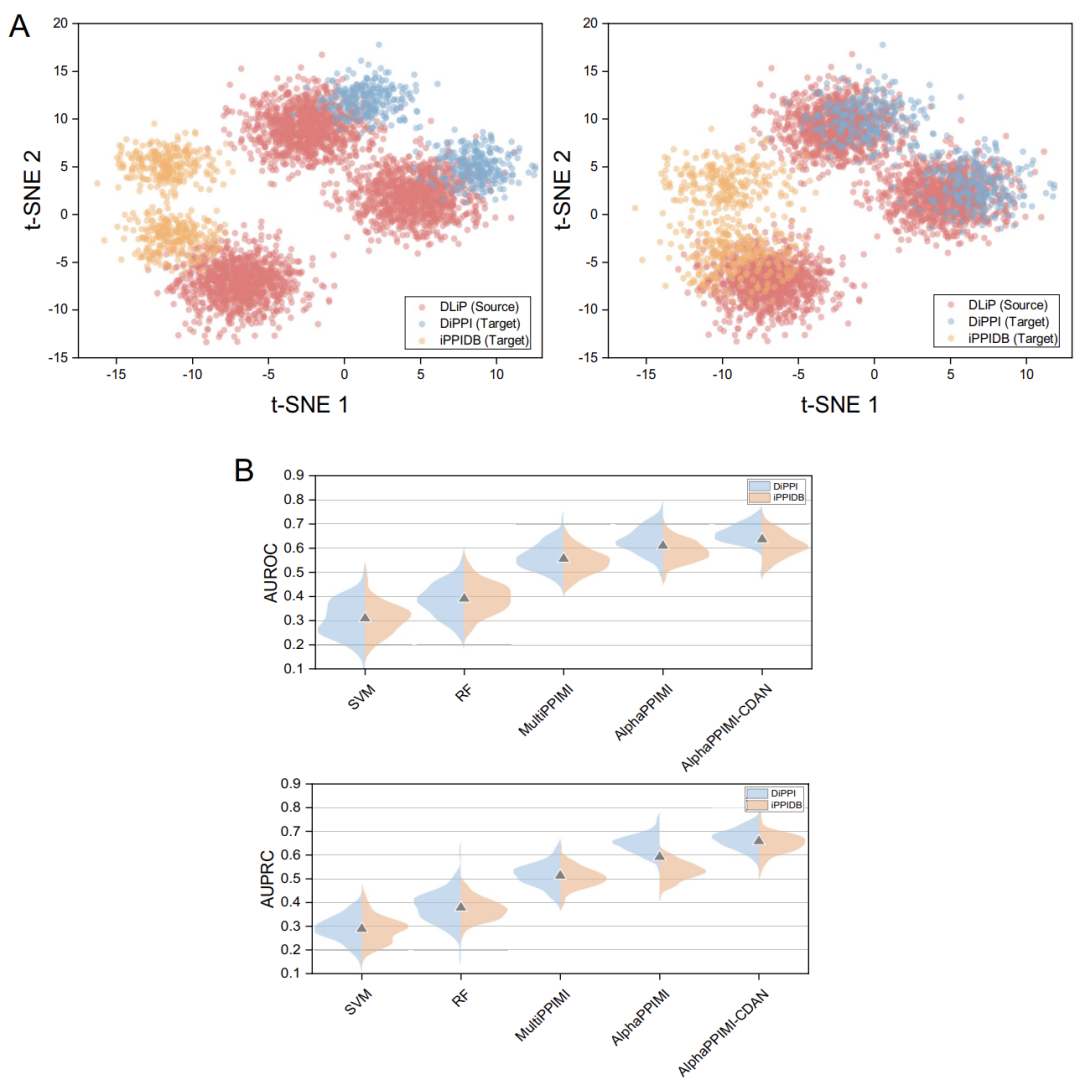
In order to test the cross-domain adaptability of AlphaPPIMI in predicting PPIs regulators, the research team designed a migration experiment.The DLiP dataset is regarded as the “source domain” (data used for model training), and the DiPPI and iPPIDB datasets are regarded as the “target domains” (data used for model validation).
Experimental results show that the performance of all models degrades under domain shift, but AlphaPPIMI is more robust.Its AUROC and AUPRC on DiPPI are significantly higher than those of MultiPPIMI. The gap between cross-domain and intra-domain performance highlights the necessity of domain adaptation strategies. As shown in the figure below, the study further proposed the AlphaPPIMI-CDAN architecture, which achieves cross-domain distribution adaptation through conditional feature alignment. This model comprehensively outperforms the baseline models on DiPPI and iPPIDB. Unlike traditional edge alignment methods, this method guides feature alignment based on the category conditional distribution, generating more discriminative representations. It can effectively address the distribution shift caused by subtle functional differences in PPI domains, while mitigating negative transfer to improve cross-domain prediction robustness and generalization.
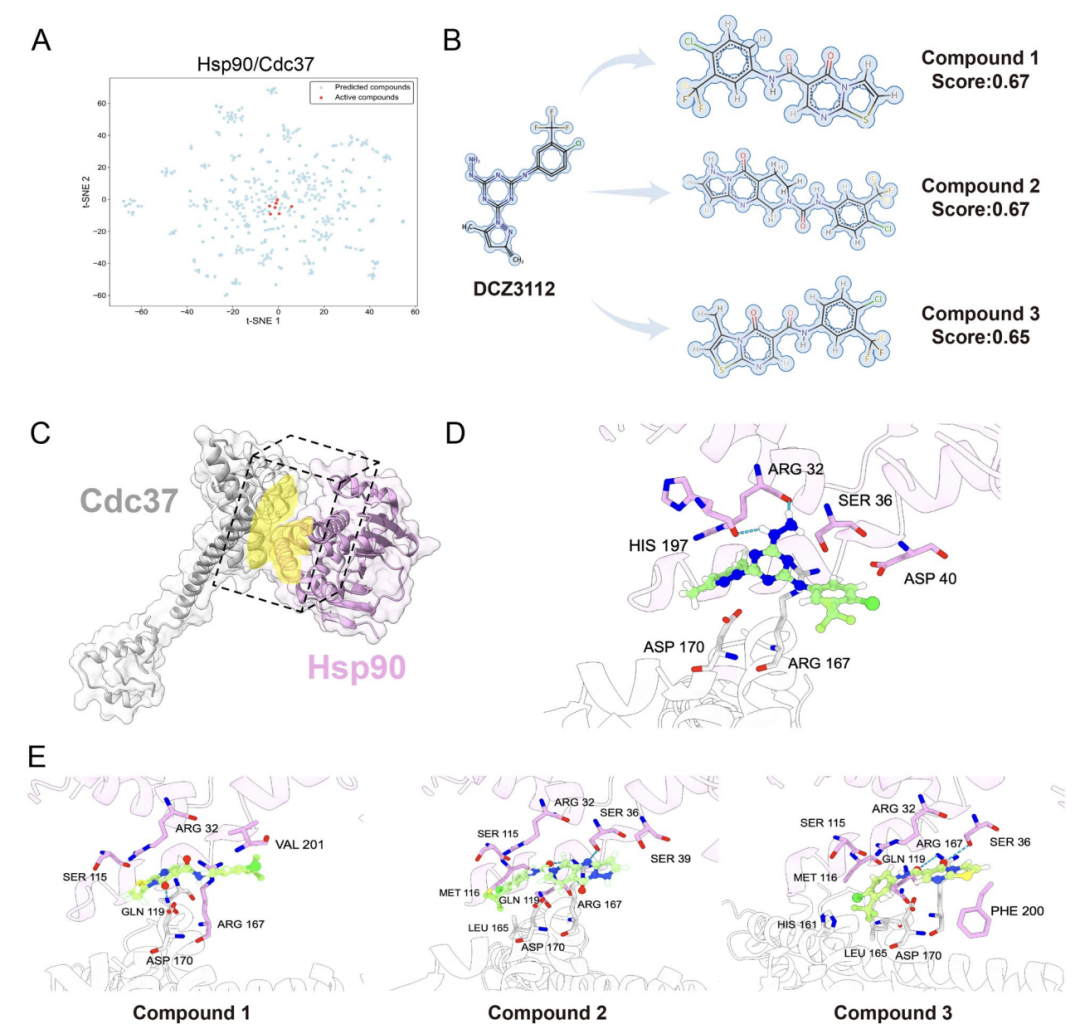
In practical application verification, the study also took Hsp90-Cdc37 PPIs, which has a clear interface structure and is a key anti-cancer target, as the object.As shown in Figure A below, AlphaPPIMI screened compounds with prediction scores > 0.8 in the ChemDiv library, whose chemical space was close to the distribution of known active inhibitors; as shown in Figure B below, researchers used the verified inhibitor DCZ3112 as a reference and screened out three candidate compounds through structural similarity and pharmacophore analysis; as shown in Figures D–E below, molecular docking showed that these compounds can form interactions similar to reference molecules with key residues such as Arg32 and Ser36, enhancing their inhibitory potential.
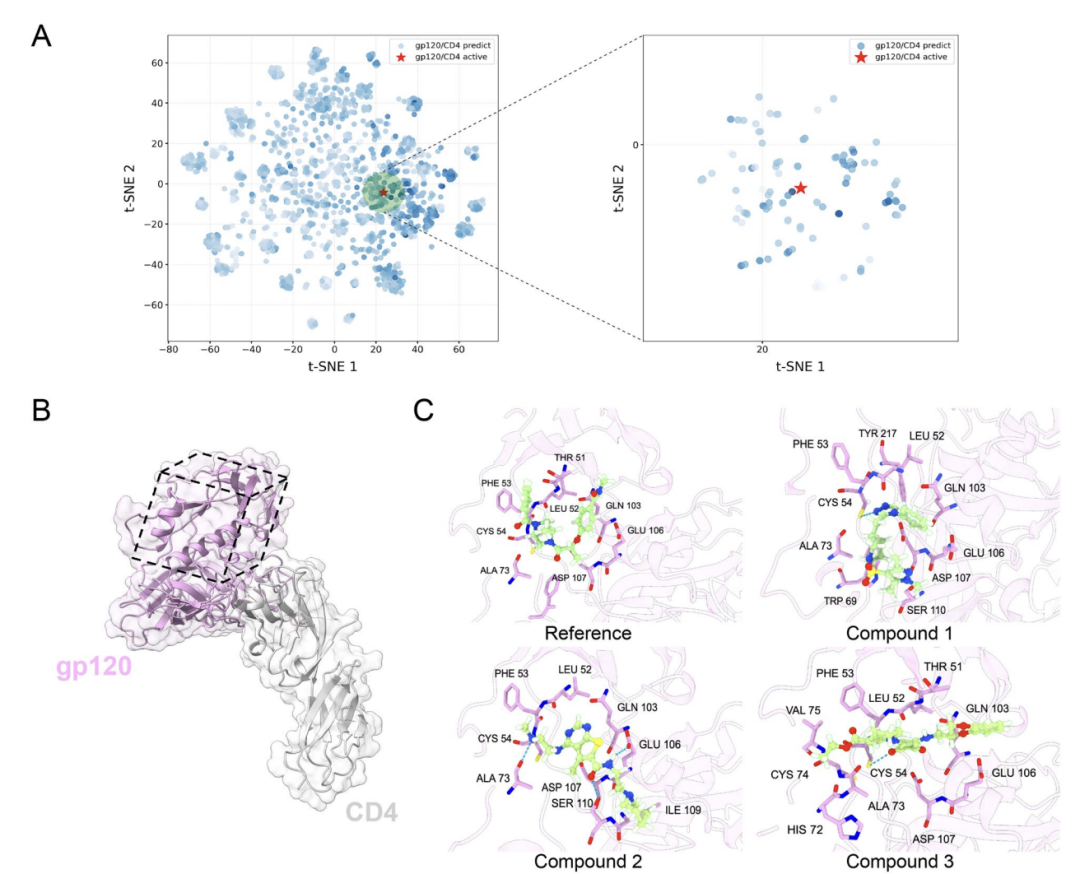
Regarding the application of AlphaPPIMI in screening allosteric PPIs modulators, as shown in Figure A below, researchers took the interaction between HIV-1 gp120 and CD4 as an example. AlphaPPIMI screened out compounds with a predicted probability greater than 0.8, whose chemical space overlapped highly with known active inhibitors. As shown in Figure BC below, molecular docking results based on the atypical interface structure (PDB: 6L1Y) showed that the candidate compounds could interact with key residues such as THR51, LEU52, and PHE53, indicating that AlphaPPIMI can discover allosteric inhibitors targeting the interface of difficult-to-druggable PPIs, providing a new approach for the development of related drugs.
Industry-university-research collaboration promotes PPIs targeted drugs from basic research to clinical application
In the research and development of PPIs targeted drugs, academia and industry are working closely together to gradually push basic research in this field towards clinical application.
At the academic frontier, many research teams are exploring more accurate and efficient PPIs prediction and targeting methods.For example, a team from Stanford University has developed a general biomedical AI agent called Biomni.This intelligent agent is capable of autonomously completing complex research tasks across multiple biomedical disciplines, including genetics, genomics, microbiology, pharmacology, and clinical medicine. The creation of Biomni marks a transition for AI in biomedical research from a tool user to an autonomous decision-maker. By integrating dispersed scientific research resources into actionable intelligent agent-based behavioral units, it not only overcomes the fragmented bottlenecks of traditional research processes but also potentially fosters the emergence of a cross-disciplinary, high-throughput, autonomous scientific discovery engine.
Another representative studySun Yat-sen University proposed a PPIs prediction method based on fusion feature extraction and a new unsupervised feature selection mechanism.Extensive experiments demonstrated that the proposed method performed well on five datasets covering both intra- and interspecies interactions, significantly outperforming 16 existing machine learning methods. This research not only provides an efficient and reliable framework for large-scale PPI prediction tasks but also demonstrates broad functional adaptability, offering a new solution for drug-drug and drug-food interaction prediction.
When it comes to industrial translation, companies are also actively advancing these academic breakthroughs into the clinic. For example, AN2025 (generic name buparlisib), developed by the Chinese biopharmaceutical technology company Adlai Nortye based on a global license from Novartis, is a pan-inhibitor specifically targeting the PI3K signaling pathway. It has now entered a global Phase III clinical trial, primarily for the treatment of patients with recurrent or metastatic head and neck squamous cell carcinoma who have progressed despite anti-PD-1/PD-L1 therapy.
Another example is Iqirvo (elafibranor), launched by the well-known French pharmaceutical company Ipsen. As the first new PBC treatment approved in the past decade, it has verified the clinical value of PPI regulation in the non-tumor field and provided a new treatment paradigm for complex metabolic diseases. Its approval has also promoted in-depth exploration of the PPAR family protein interaction network.
The deep collaboration between academia and industry in the field of PPI-targeted drugs has not only accelerated the translation of scientific research findings into clinical value but also significantly improved the efficiency and success rate of new drug development. From multimodal AI predictive models to drug candidates with clear clinical benefits, this cross-disciplinary collaboration is redefining the path of biomedical innovation. In the future, with the integration of more data and algorithms, and the deepening of cross-institutional and interdisciplinary collaboration, PPI-targeted drugs may bring even more breakthroughs in the treatment of complex diseases.
Reference Links:
1.https://mp.weixin.qq.com/s/ryYJ6T7qEjnjvkhBL4-dAA
2.https://mp.weixin.qq.com/s/7upIPYam1LR0TiGBYXmkOw
3.https://mp.weixin.qq.com/s/69GU1R5lXHdTLttlT8apyw








