Command Palette
Search for a command to run...
The MIT Team Proposed the FASTSOLV Model, Which Is 50 Times Faster Than the Original Model, to Predict the Solubility of Small Molecules at Any temperature.

In the fields of chemistry and materials science, the solubility of organic solids in different solvents is a core molecular property, impacting the entire research and industrial chain. For synthetic processes, precise control of solubility not only helps screen the optimal solvent and optimize reaction conditions, but also significantly improves product yield and purity, reducing production costs. In environmental science, it is a key parameter for analyzing the migration and fate of pollutants such as per- and polyfluoroalkyl substances (PFAS) in soil and water, providing a scientific basis for pollution prevention and control. And in processes such as crystallization and membrane separation, solubility is a core variable that determines phase behavior and separation efficiency.
However, traditional experimental determination methods have many limitations: they are not only time-consuming and material-consuming, but are also easily interfered with by factors such as organic solid crystal form and impurities, resulting in insufficient data accuracy. According to research, the inter-laboratory standard deviation of water solubility logS is often as high as 0.5–0.7 log units, and in extreme cases, the difference in measurement results can even exceed 10 times. Although empirical group addition methods, quantum chemical models, and machine learning methods have been applied to prediction,However, there are often problems with insufficient versatility or difficulty in balancing accuracy and computational efficiency.
To address this pain point, a research team from the Massachusetts Institute of Technology combined chemical informatics tools with the new organic solubility database BigSolDB.Improved on the basis of FASTPROP and CHEMPROP model architecture,The model can simultaneously input solute molecules, solvent molecules and temperature parameters, and directly perform regression training on logS.
In a strict solute extrapolation scenario, compared with existing SOTA models such as Vermeire,The RMSE of the optimized model was reduced by 2–3 times, and the inference speed was increased by up to 50 times.Currently, the team has named the FASTPROP derivative model FASTSOLV and has released it as open source, providing an efficient and practical tool for related scientific research and industrial applications.
The relevant research results were published in Nature Communication under the title "Data-driven organic solubility prediction at the limit of aleatoric uncertainty."

Paper address:
https://www.nature.com/articles/s41467-025-62717-7
Follow the official account and reply "Organic Solubility" to get the full PDF
BigSolDB-driven dataset construction and evaluation system design
The core data source of this study is BigSolDB, which systematically collects solubility data of organic solids in a variety of organic solvents and under different temperature conditions close to the precipitation limit, providing key support for the training of general prediction models.
To achieve the research goal of “extrapolating new solutes without any prior knowledge,” the research team designed a rigorous training-evaluation system:The model was trained on BigSolDB and independently tested on two public datasets: SolProp and Leeds.To avoid underestimation of the difficulty of extrapolation, as shown in the figure below, this study first removed all solutes in SolProp that overlapped with BigSolDB, and introduced the Leeds dataset with a wider chemical space as a supplement.
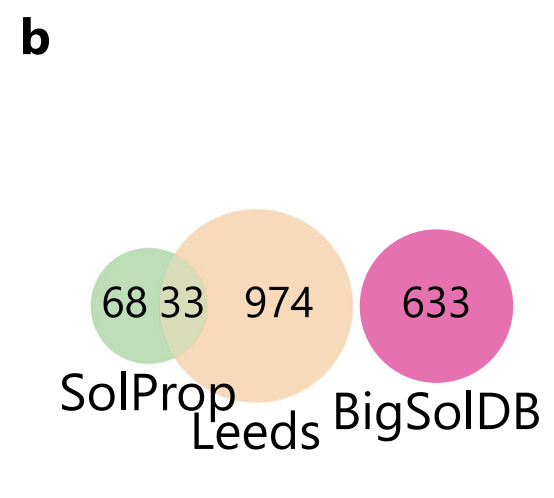
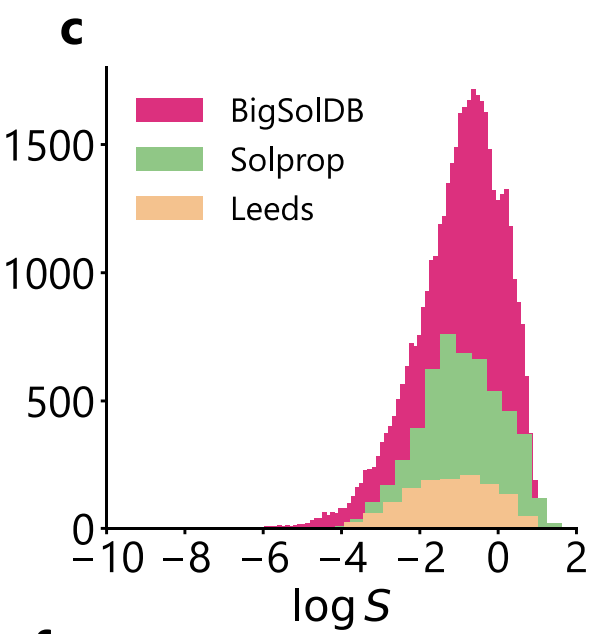
Compared with SolProp,Leeds provides a higher solute diversity but only covers room temperature conditions.This not only allows for testing the model's adaptability to new chemical spaces, but also provides a higher upper uncertainty limit due to the lack of implicit noise reduction from "multi-temperature averaging." Notably, as shown in the figure below, the logS distributions for the three datasets are highly consistent, all concentrated near –1 and exhibiting a long tail at the low solubility end, ensuring distributional comparability for performance comparisons across datasets.
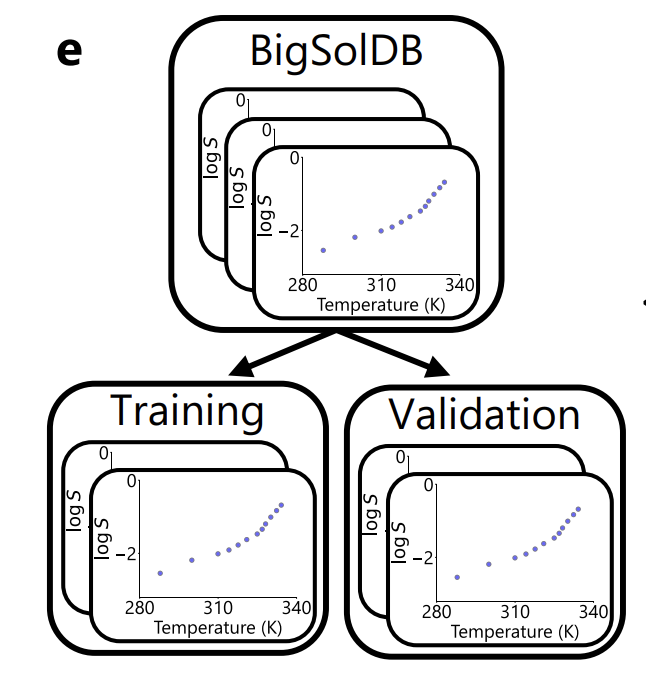
In terms of data segmentation, as shown in the figure below, the researchers strictly use solute as the unit: 95% of solute is used for training, 5% is used for validation and model selection,All measurements of the same solute in different solvents and temperatures will not appear in different subsets at the same time.This effectively avoids information leakage.
In addition, the study used the ASTARTES toolkit to randomly divide the validation set into "complete experiments" in the training data, and re-checked the division boundaries from both the solute and experimental dimensions in the final evaluation to ensure the independence and rigor of the evaluation.
FASTSOLV model construction driven by BigSolDB
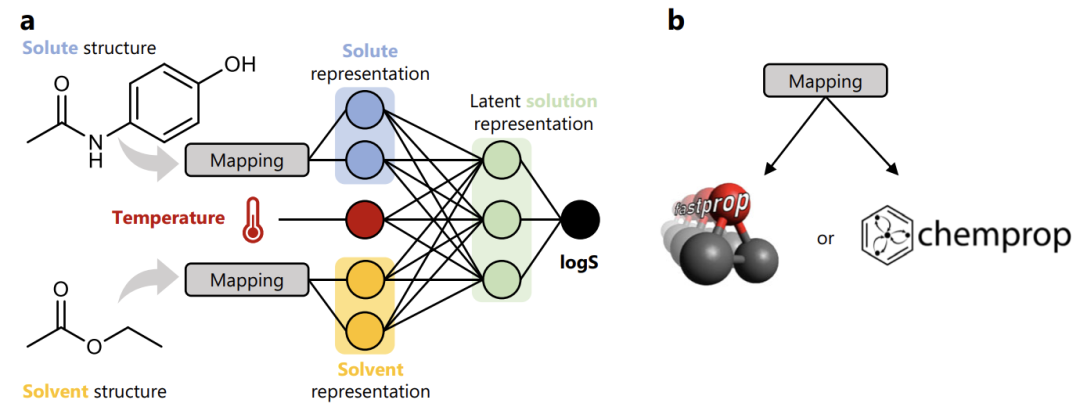
Relying on the BigSolDB dataset, as shown in the figure below, this study customized the two classic model architectures of FASTPROP and CHEMPROP and built a clear machine learning modeling process.
first,Map the molecular structures of solutes (such as paracetamol) and solvents (such as ethyl acetate) into corresponding representation vectors;Then,These two molecular representation vectors are combined with the solution temperature parameter to form a complete solution representation.final,The representation was input into a fully-connected neural network and regression training was performed with logS (logarithm of solubility) as the target.
Through this transformation, the model finally developed achieves unified prediction of small molecule solubility in multiple organic solvents + different temperature scenarios, breaking the traditional model's dependence on specific solvents or temperature ranges.
To further improve the robustness and predictive reliability of the model, the research team did not rely on a single model output.Instead, the FASTPROP model is trained under four different random initialization conditions, and then the final FASTSOLV model is obtained through the integration strategy combination.All subsequent key analyses, such as performance comparisons and case verifications, are based on this integrated model, effectively reducing the random fluctuation risk of a single model.
At the same time, to objectively measure the performance of the new model, the study introduced the currently widely recognized SOTA model, the Vermeire model, as a comparison benchmark. This model is trained through four independent thermochemical sub-models and then outputs solubility results through thermodynamic cycle combination. It has the advantage of balancing solvent diversity and temperature dependence. However, the study found that the SolProp dataset used for its testing has a large amount of solute structure overlap with its own training set. This "data overlap" may lead to an overestimation of the extrapolated performance. To ensure fairness and rigor in the comparison, this study strictly reproduced the original training-testing setup of the Vermeire model and conducted control experiments on this basis to ensure that the performance difference is only due to the model itself and not the test conditions.
Updates organic solubility extrapolation SOTA with 2–3 times greater accuracy and 50 times greater speed
This study conducted multi-dimensional testing and verification of the model performance. In the interpolation scenario, the optimized FASTPROP model achieved RMSE=0.22, P₁=94%, and the CHEMPROP model achieved RMSE=0.28, P₁=90%.The performance has approached the noise ceiling of experimental data, confirming the supporting value of BigSolDB.
In the new solute extrapolation test, as shown in the figure below, the Vermeire model performed poorly on the Leeds dataset due to systematic overestimation (RMSE=2.16, P₁=34%), while the RMSE of FASTPROP and CHEMPROP dropped to 0.95 and 0.99, respectively, and P₁ exceeded 69%. On the SolProp dataset, our model also performed better (RMSE=0.83, P₁=80%).And the inference speed of FASTPROP is about 50 times that of the Vermeire model.Supports SHAP interpretability analysis.
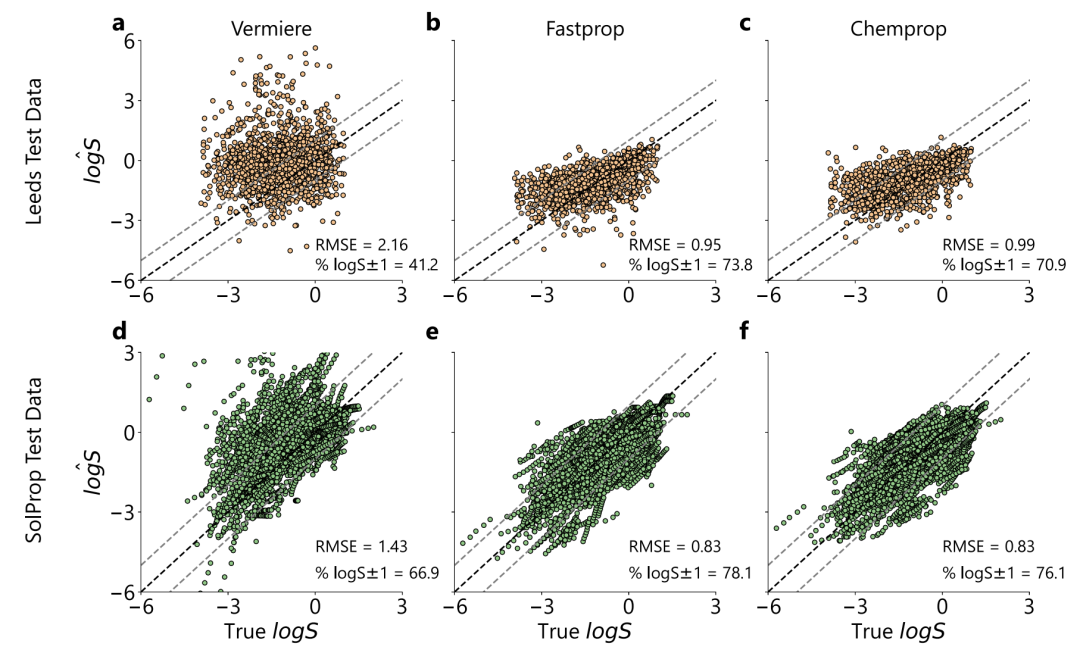
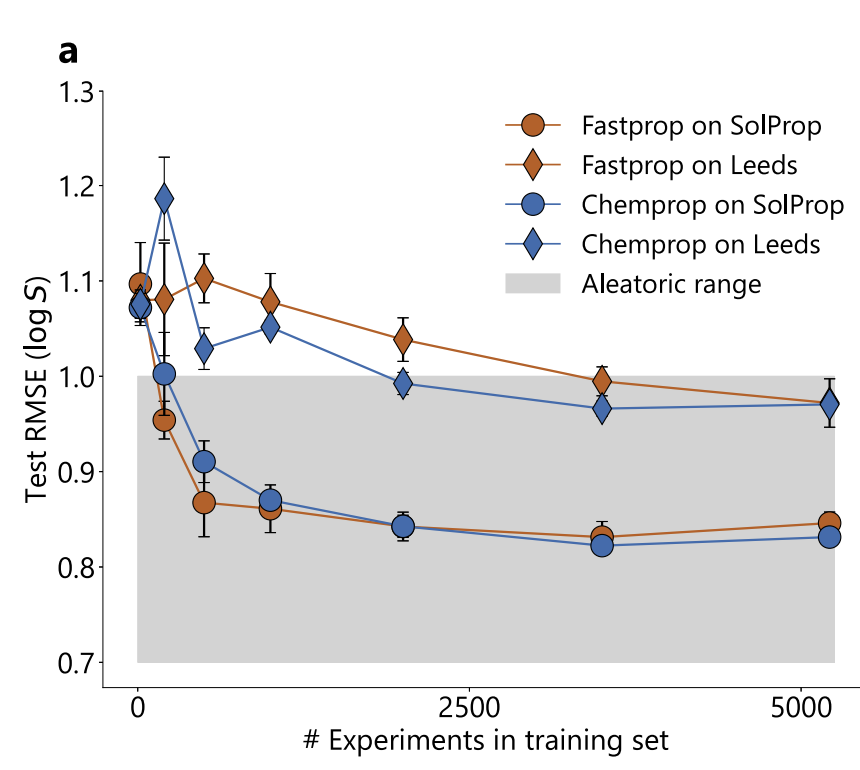
The training data volume experiment is shown in the figure below. Although FASTPROP and CHEMPROP have different molecular representations, their performance converges to similar limits: the SolProp test set requires about 500 experiments (≈5,000 data points) to reach the plateau, while CHEMPROP requires about 2,000 experiments (≈20,000 data points) on the Leeds test set.
Estimated from 34 sets of multi-source data under the same conditions in BigSolDB, the experimental random uncertainty limit is RMSE=0.75 log units, while the RMSE of the two models on SolProp is 0.83, which is close to this limit; compared with large models such as MolFormer and ChemBERTa-2, the two models perform better.It proves that the performance bottleneck comes from experimental data rather than model expressiveness.
Mean test of model performance at arbitrary limits
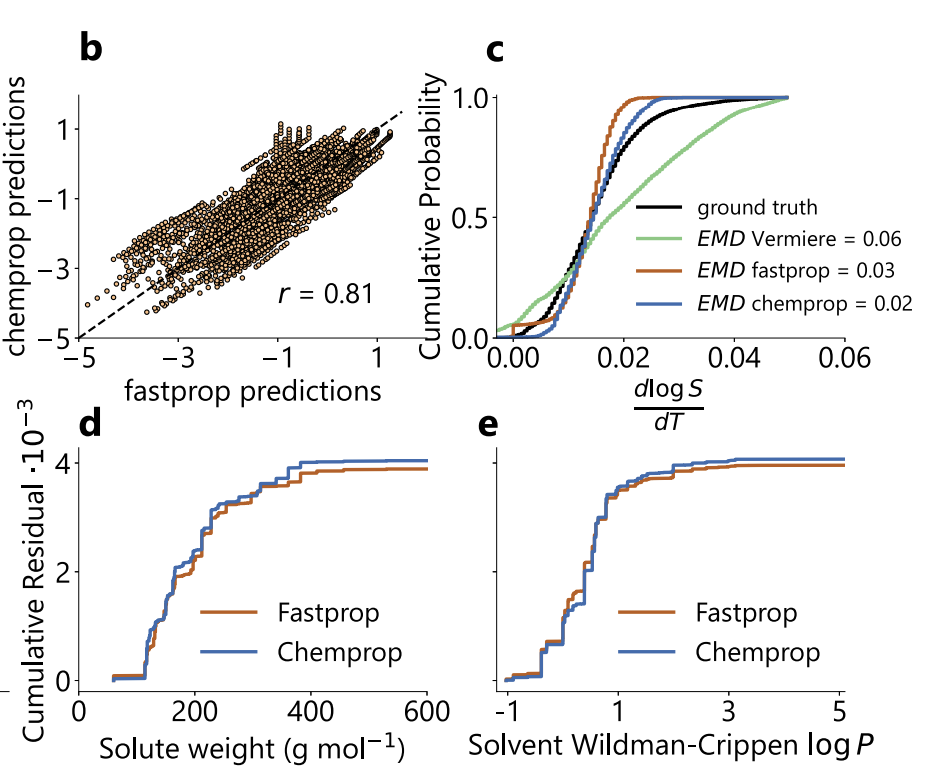
In addition, as shown in the figure below, the two models have highly correlated predictions on the SolProp test set (Pearson r=0.81), and the predicted temperature gradient distributions are also highly consistent (EMD=0.03/0.02). The systematic error is significantly lower than that of the Vermeire model (EMD=0.06).
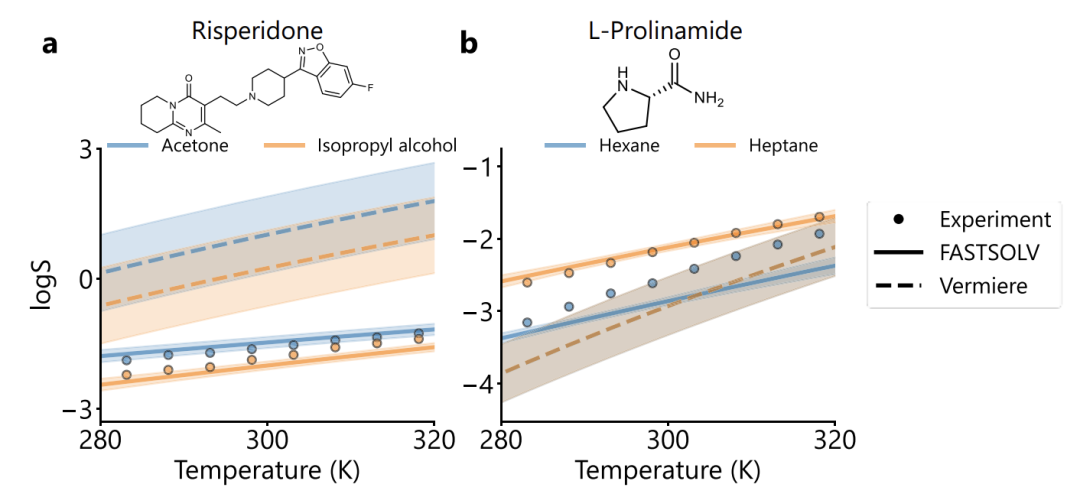
The study also found that in the typical solute validation, as shown in the figure below, FASTSOLV has a significant advantage in predicting risperidone (RMSE=0.16 vs Vermeire 1.64) and L-proline (RMSE=0.25 vs Vermeire 2.33).Not only can it correctly determine the order and temperature dependence of solvent solubility, but it can also distinguish between hexane and heptane, which have similar structures.Failure mode analysis showed that the prediction error of anthraquinones was high, but in the subset of 85 anthraquinone/anthraquinone derivatives, the overall RMSE of the model was 0.52, and the solvent solubility could be stably ranked, indicating that the molecular characterization was reasonable.
In summary,Compared with the Vermeire model, FASTSOLV reduces RMSE by 2–3 times and accelerates inference by up to 50 times.This method combines interpretability with engineering potential, representing state-of-the-art performance under strict extrapolation settings. The study also points out that adding additional training data will not break through performance limits, and future research will focus on building a high-precision organic solvent dataset.
"Dataset + AI" drives global breakthrough in molecular property prediction
In today's wave of cross-innovation in chemistry, medicine, and materials science, molecular property prediction technology, centered on "large-scale data sets + advanced machine learning models," is becoming a key tool for addressing industry pain points such as time-consuming experiments, high R&D costs, and difficult performance prediction.
In academia, research teams around the world are responding to the breakthroughs of FASTSOLV and BigSolDB by launching a series of innovative solubility prediction studies. For example, researchers at the University of Leeds in the UK proposed a Causal Structure Property Relationship model that combines artificial intelligence with physical-chemical mechanisms.The solubility prediction in organic solvent and water system is almost as accurate as the experimental error.It also has outstanding interpretability and is considered an important milestone in the field of solubility modeling.
Meanwhile, a research team at the Massachusetts Institute of Technology (MIT) has made significant progress in antibiotic discovery using the graph neural network Chemprop. They determined the antibiotic activity and human cytotoxicity profiles of 39,312 compounds and used graph neural network ensembles to predict the antibiotic activity and cytotoxicity of 12,076,365 compounds for new antibiotic discovery. By screening a panel of initial compounds and evaluating their growth inhibitory activity against the methicillin-sensitive strain S. aureus RN4220,512 active compounds were obtained.The graph neural network is then trained to perform binary classification predictions.
In the pharmaceutical industry, remarkable innovations are also emerging. The pharmaceutical industry has long focused on high-throughput, low-cost solubility assessment technologies. For example, AspenTech's Aspen Solubility Modeler tool can predict solubility in hundreds of solvent combinations based on measured data in a few solvents. This tool significantly improves efficiency and decision-making reliability in crystal screening and process development at major companies like GSK and AstraZeneca.
In addition, some companies are leveraging similar data-driven models in the field of materials research and development. By analyzing large amounts of molecular structure and performance data, they are predicting the properties of new materials, shortening R&D cycles, and reducing R&D costs. In the chemical industry, some companies are using models to predict the effects of chemical reactions under different solvents and temperature conditions, optimizing production processes and improving production efficiency and product quality. These are all examples of companies applying models and data concepts from academic research to actual production innovation.
Reference Links:
2.https://www.manufacturingchemist.com/news/article_page/Solubility_modelling/57726
Get high-quality papers and in-depth interpretation articles in the field of AI4S from 2023 to 2024 with one click⬇️









