Command Palette
Search for a command to run...
Redefining the Classification of Protein Language Models Based on the Relationship Between Structure/Sequence/Function: Dr. Li Mingchen Explains Protein Language Models in Detail

The third "AI for Bioengineering Summer School" of Shanghai Jiao Tong University officially opened from August 8 to 10, 2025.This summer school brought together more than 200 young talents, scientific researchers and industry representatives from more than 70 universities, more than 10 scientific research institutions and more than 10 industry-leading companies around the world, focusing on the integrated development of artificial intelligence (AI) and bioengineering.
Among them, in the "Frontiers of AI Algorithms" course section, Li Mingchen, a postdoctoral fellow in the Hong Liang research group at the Institute of Natural Sciences of Shanghai Jiao Tong University, shared with everyone the cutting-edge achievements of protein language models in function prediction, sequence generation, structure prediction, etc., with the theme of "Basic Model of Proteins and Genomes", as well as related research progress in expansion laws and genome models.

HyperAI has compiled and summarized Dr. Li Mingchen's wonderful sharing without violating the original intention. The following is the essence of the speech.
A new classification of protein language models: the relationship between protein structure, sequence and function
Proteins have a wide range of applications, encompassing fields such as chemical engineering, agriculture, food, cosmetics, medicine, and testing, with a market value exceeding trillions of dollars. Simply put, protein language modeling is a probability distribution problem. It's equivalent to determining the probability of an amino acid sequence occurring in nature and sampling accordingly. Through pre-training on massive amounts of data, the model can effectively represent the probability distribution found in nature.
The protein language model has three core functions:
* The learning process of representing protein sequences as high-dimensional vectors
* Determine the rationality of the amino acid sequence
* Generate new protein sequences
Many research papers categorize protein language models by Transformer architecture, directly describing them as either Transformer Encoder or Transformer Decoder-based. This classification is difficult for biology researchers to understand and often causes confusion. Therefore, I will introduce a new classification method:Classification based on the relationship between protein structure, sequence and function.
The sequence of a protein is its amino acid sequence. Once the amino acid sequence is known, it can be synthesized in a laboratory or factory and applied in practice. The structure of a protein is equally crucial. Its function is due to its specific structure in three-dimensional space, which enables its function at the microscopic level.
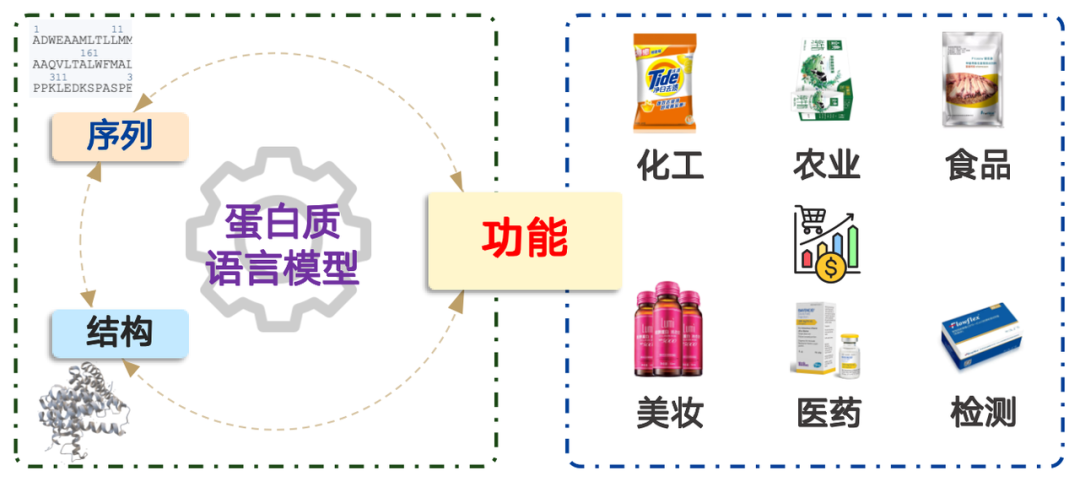
Based on this idea, protein language models can be divided into the following four categories:
1. Sequence → Function:Predicting the function of a given amino acid sequence, i.e. Functional prediction model.
2. Function → Sequence:Design the corresponding amino acid sequence according to the given function, including Generative Modelsand Mining Model.
3. Sequence → Structure:Predicting its structure based on its amino acid sequence is usually called "Structural prediction model",The Nobel Prize-winning AlphaFold belongs to this type of model.
4. Structure → Sequence:Designing a corresponding sequence based on a given protein structure is usually called "Reverse folding model".
Application scenarios and technical paths: Analysis of four mainstream models
Sequence → Function
The simplest way to understand "sequence → function" is supervised learning.
First, the most basic function prediction model involves expressing protein sequences as vectors and then training them on a specific dataset. For example, if we want to predict protein melting points, we first need to collect a large amount of protein melting point labels, convert all protein sequences in the training set into high-dimensional vectors, and train them using supervised learning methods. Finally, we can perform inference on sequences in the test or prediction set to predict function. This approach can handle a wide range of tasks and is currently a hot research topic, as well as one that is relatively easy to produce results.
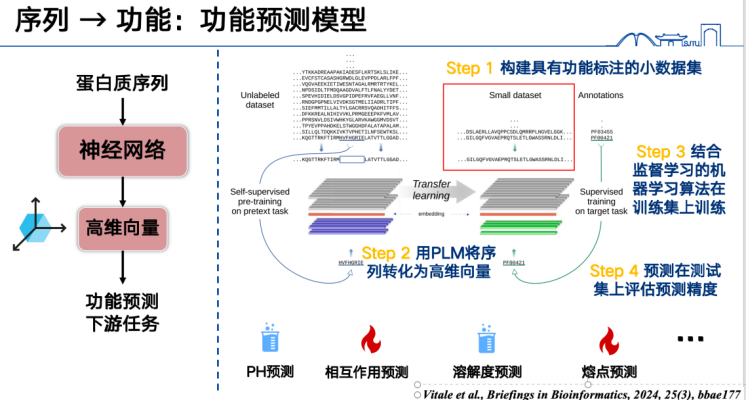
Secondly, the protein language model can also predict mutation functions.The core idea is to make some changes to certain amino acids in the protein sequence, and then use the protein language model to determine whether the change is "reasonable".
The "reasonable" here doesn't refer to logical conformity in real life, but rather to whether the amino acid change conforms to the probability distribution of natural protein sequences. This probability distribution comes from a large number of real amino acid sequence statistics, and these amino acid distributions are themselves the product of tens of millions of years of evolution.
The protein language model learns these evolutionary laws during training and can therefore determine whether a mutation conforms to or deviates from these laws. Mathematically, this determination can be converted into the ratio of the probabilities of the two sequences before and after the mutation. For ease of calculation, this ratio is often logarithmized, converting it into a subtraction form.
The likelihood ratio between mutants and wild-type proteins used by language models can estimate the strength of a mutation's effect. This idea was first demonstrated in a 2018 Nature Methods paper introducing the DeepSequence model, but the model was relatively small at the time. Subsequently, in 2021, the ESM-1v model further demonstrated that protein language models can also effectively predict mutation effects using likelihood ratios.
To evaluate the accuracy of the protein mutation function prediction model, a benchmark is needed.
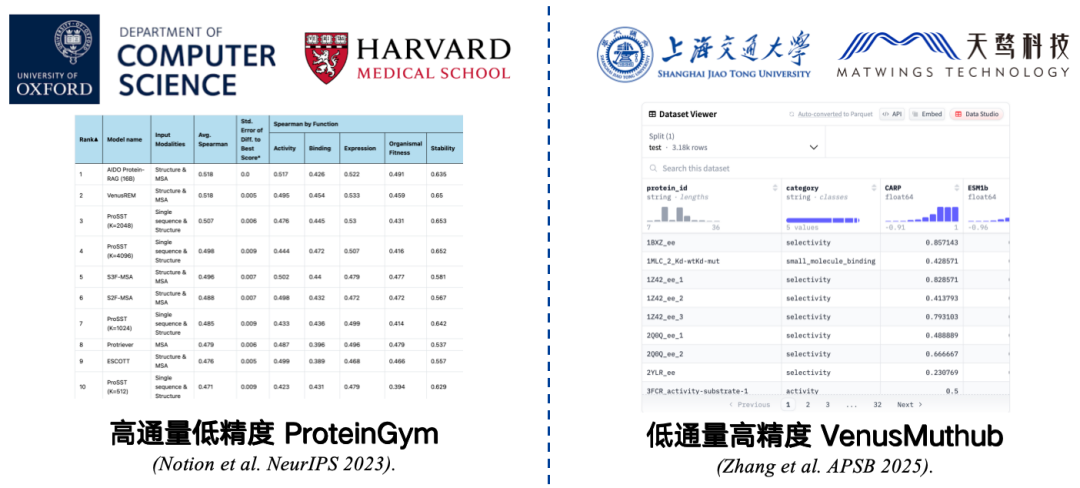
Benchmarks are small sets of data collected to measure accuracy. For example, ProteinGym, jointly developed by Harvard Medical School and the University of Oxford, is the most commonly used benchmark. It contains data on 217 mutant proteins and millions of mutation sequences. Researchers assign scores to each of these mutation sequences using a protein language model, then compare the model's predicted scores with the actual scores. A higher correlation indicates better model performance.
However, ProteinGym is a high-throughput, low-precision benchmark.While limited by experimental conditions, it can be tested on a large scale, the accuracy may be limited. Repeating an experiment may result in errors in the correlation between the results and the original data, making the evaluation results inaccurately reflect the performance of the model in real-world applications.
To solve this problem,We have developed a low-throughput, high-precision small sample benchmark such as VenusMutHub.Although the amount of data is not large, each piece of data is relatively accurate, and the results of repeated experiments are almost consistent, which is closer to real application scenarios.
* Paper address:Zhang L, Pang H, Zhang C, et al. VenusMutHub: A systematic evaluation of protein mutation effect predictors on small-scale experimental data[J]. Acta Pharmaceutica Sinica B, 2025, 15(5): 2454-2467.
Furthermore, structure can be introduced to enhance the mutation prediction accuracy of protein language models. Last year, our team published a paper on a protein language model, the ProSST model, at NeurIPS. This model uses both amino acid sequences and structured sequences to perform multimodal pre-training. ProSST ranked first on the ProteinGym Benchmark, the largest zero-shot mutation prediction benchmark.
* Paper address:Li M, Tan Y, Ma X, et al. ProSST: Protein language modeling with quantized structure and disentangled attention[C]. Advances in Neural Information Processing Systems, 2024, 37: 35700-35726.
When doing experiments or designs, you often encounter questions like: "Which model should I use?" and "How should I choose as a user?"
In a study published this year,Our team found that the perplexity of the protein language model for the target sequence can roughly reflect its accuracy in the mutation prediction task.The advantage is that it can provide a performance estimate without requiring any target protein mutation data. Specifically, the lower the perplexity, the better the model's understanding of the sequence, which often means that its mutation predictions for that sequence will be more accurate.
Based on this idea, we developed an ensemble model, VenusEEM. It weights models based on perplexity, or directly selects the model with the lowest perplexity. This improves mutation prediction accuracy to a high standard. Regardless of the strategy, the final prediction score remains relatively stable, preventing significant performance degradation due to incorrect model selection.
* Paper address:Yu Y, Jiang F, Zhong B, et al. Entropy-driven zero-shot deep learning model selection for viral proteins[J]. Physical Review Research, 2025, 7(1): 013229.
Finally, in the "sequence to function" research direction, in addition to the previously mentioned models, our team also developed a novel iterative high-site mutation design model, PRIME, last year. Specifically, we first pre-trained a large protein language model on 98 million protein sequences. For the high-site mutation prediction task, we first obtained low-site mutation data and input it into the protein language model, encoding it into a function vector. Based on this function vector, we then trained a regression model to predict high-site mutations.Through this iterative reaction, an excellent protein product can be developed in just 2-3 rounds of experiments.
* Paper address:Jiang F, Li M, Dong J, et al. A general temperature-guided language model to design proteins of enhanced stability and activity[J]. Science Advances, 2024, 10(48): eadr2641.

「Function→Sequence」
What we have discussed before is from sequence to function. Let’s think about whether we can reversely deduce the sequence from the function?
There's a forward and inverse problem between sequences and functions. While the forward problem is about finding a definitive answer, the inverse problem involves searching for a solvable solution within a vast feasible space. Generating sequences from functions is precisely this inverse problem. The reason for this is that sequences typically correspond to only one or a few functions, but a single function can be implemented by a variety of completely different sequences. Furthermore, there's no reliable benchmark for the inverse problem. When a model generates sequences from a given function, its accuracy can usually only be tested experimentally.

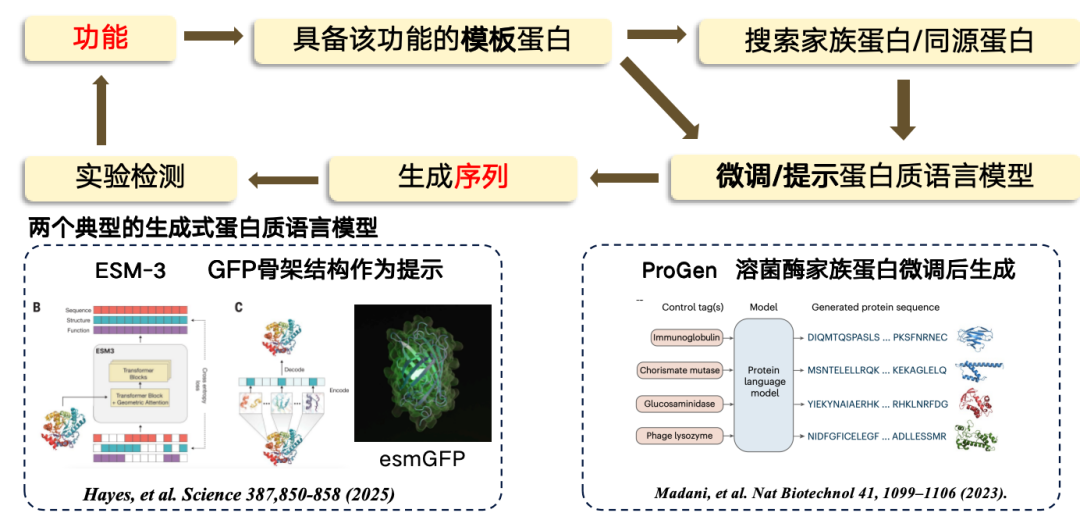
In the current study,The design from function to sequence mostly adopts a template-based approach. If a template protein is known to have a specific function, it can be used as a basis to find or generate a new region.The process is to first go from function to template sequence, and then search out some family proteins/homologous proteins from the template protein, and then fine-tune the protein language model, use the fine-tuned language model to generate new sequence regions, and finally conduct experimental testing.
Currently, the two most representative generative protein language models include:
*ESM-3, produced using green fluorescent protein (GFP) as a template, but the resulting protein is less functional.
* ProGen is a purely autoregressive language model, similar to ChatGPT, that can be generated based on functional cues. It is generated by fine-tuning the protein architecture of lysozyme.
In addition to directly generating new protein sequences,You can also search directly from the massive amount of existing protein sequences.The template protein is encoded in a high-dimensional space, and the distance between the vectors determines whether the two proteins have the same function. Finally, the results are retrieved from a database. The principle behind this approach is that the distance between the encodings or vectors of two proteins in a high-dimensional space can roughly reflect whether the two proteins have similar functions.
The figure below illustrates two typical examples of protein language model mining. The first is ESM-Ezy, developed by Westlake University, which uses the ESM-1b model to perform vector searches and mine multiple expressions for infilling. The second is the VenusMine large-scale model, which mines highly efficient PET hydrolases.
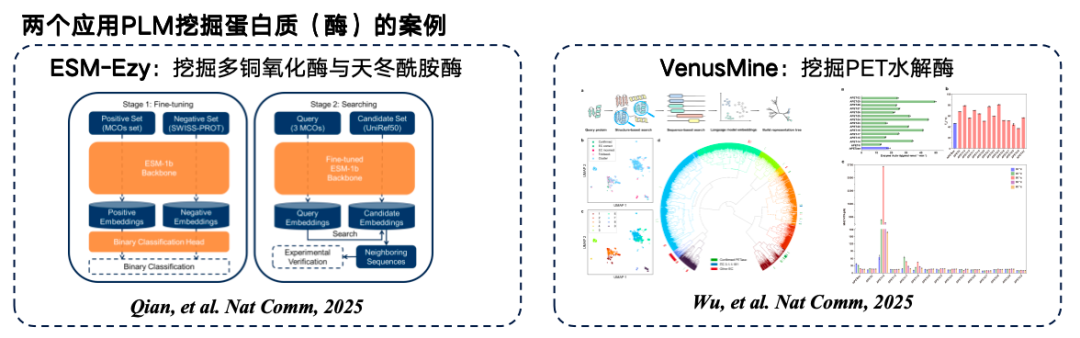
* Paper address:Wu B, Zhong B, Zheng L, et al. Harnessing protein language model for structure-based discovery of highly efficient and robust PET hydrolases[J]. Nature Communications, 2025, 16(1): 6211.
In addition to "function → sequence", you can also add "mediator" between function and sequence:
* When structure is used as an intermediary: protein structure is inferred based on function (common tools such as RFdiffusion), and the generated structure is then input into an inverse protein folding language model (such as ProteinMPNN) to finally generate a sequence.
* When natural language is used as a medium: For example, the method described in the research paper "A text-guided protein design framework" aligns natural language and protein sequences into a high-level space through comparative learning. Then, a protein sequence can be generated directly in this high-level space using natural language guidance.
Sequence → Structure
In the sequence-to-structure direction, the most classic model is undoubtedly AlphaFold. So, why do we still need protein language models for structure prediction?The main reason is - fast.
The main reason for AlphaFold's slowness is that its MSA (multiple sequence alignment) search relies on the CPU to search large databases. While GPU acceleration is possible, the actual acceleration is even slower. Secondly, AlphaFold also requires template matching during the folding process, which also consumes a significant amount of time. Replacing these two modules with a protein language model can greatly accelerate the structure prediction process. However, according to currently published research, the accuracy of structure prediction based on protein language models is still generally lower than that of the AlphaFold model on most evaluation metrics.
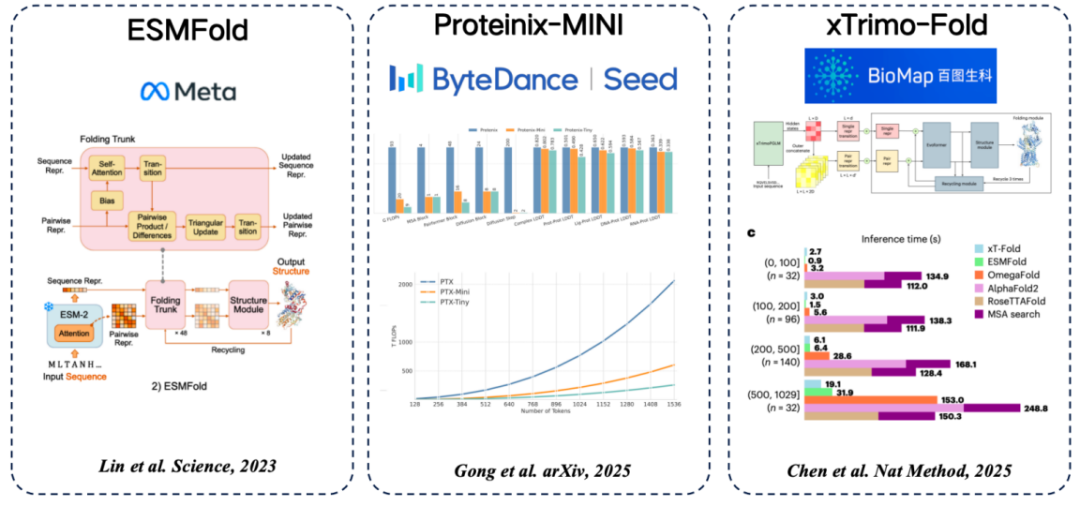
There are several common protein language models from sequence to structure.The common idea of using features extracted from protein language models to replace MSA is adopted:
* ESMFold (Meta): The first method to directly predict protein structure using a protein language model, achieving high accuracy without relying on MSA search.
* Proteinix-MINI (ByteDance): uses the protein language model instead of MSA, which also achieves very fast results and has prediction accuracy close to the AlphaFold 3 model.
* xTrimo-Fold (Baidu Biosciences): It uses the features of a model with hundreds of billions of parameters instead of MSA, which speeds up the search.
Structure → Sequence
The structure is designed based on known functions, but how to synthesize it in the laboratory?We also need to convert it into a sequence of amino acids, which is the "inverse folding language model" mentioned earlier.
The inverse folding language model can be thought of as the "inverse problem" of AlphaFold. Unlike AlphaFold, which predicts 3D structure from amino acid sequences, the inverse folding model aims to learn a mapping function from a protein's 3D structure to its amino acid sequence.
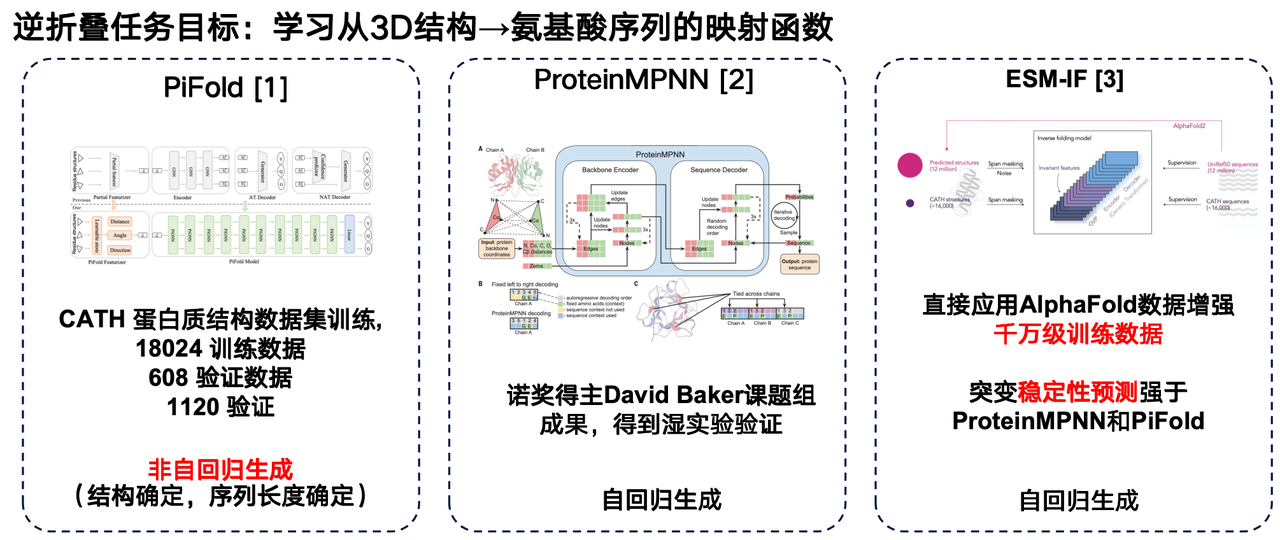
I would like to share several works in this field: The first work is the PiFold model from the Westlake University research team. A major innovation of its architecture is the use of non-autoregressive generation method.
The second is ProteinMPNN developed by David Baker's research group. As one of the most widely used inverse folding models, it uses autoregressive generation to encode individual protein structures through graph neural networks and then generate amino acid sequences one by one.
Meta's ESM-IF is also a significant advancement. Its highlight lies in leveraging the massive structural data predicted by AlphaFold to uniformly predict the corresponding three-dimensional structures for tens of millions of protein sequences, thereby constructing an extremely large training set. ESM-IF's training data reaches tens of millions of units, and the model count exceeds 100 million parameters. Based on this, the model not only performs reverse folding tasks but also demonstrates strong performance in predicting mutational stability.
Multiple approaches to enhance protein language models
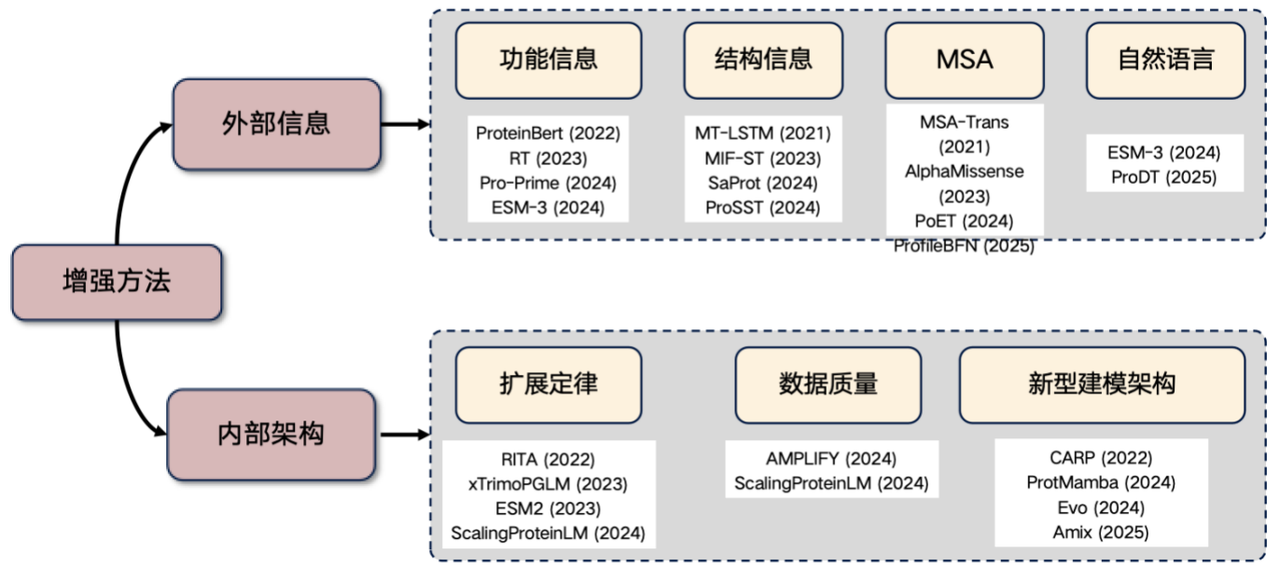
Finally, let me add a very popular research direction at present - the enhancement of protein language model. If you plan to conduct research in this field, you can start from the following ideas:Introduce external information and improve internal structure.
1. Introducing external information
* Functional information: For example, inputting features such as temperature and pH into the Transformer. This information can be incorporated into the model input explicitly or through learned actions to enhance the performance of the protein language model.
* Structural information: Introducing three-dimensional structure or structured sequence information.
* MSA information: Multiple sequence alignment (MSA) is a very useful type of information. Introducing it into the language model can often significantly improve performance.
* Natural language information: In recent years, some studies have attempted to incorporate natural language information, but this direction is still under exploration.
2. Improve internal architecture
* Scaling Law: Performance improvement is achieved by significantly increasing the number of model parameters and the size of training data.
* Improve data quality: reduce noise in data and improve accuracy.
* Exploration of new architectures: such as CARP, ProtMamba, and Evo architectures.
In recent years, using protein structure information to enhance model performance has become a hot research direction.
One of the earliest representative studies is the 2021 paper "Learning the protein language: Evolution, structure, and function," which demonstrated how structural information can be used to enhance the capabilities of protein language models. Subsequently, the SaProt model proposed a clever approach: it concatenates the protein's amino acid vocabulary with 20 virtual structure vocabularies generated by Foldseek for protein structures, ultimately generating a combined vocabulary of 400 (20 × 20) words. This vocabulary was used to train a masked language model, achieving excellent accuracy.
Our team also independently trained a multimodal pre-training model ProSST for protein sequence and structure. This model achieves a discrete representation of structural information by converting the continuous structure of the protein into discrete tokens (2,048 different tokens).
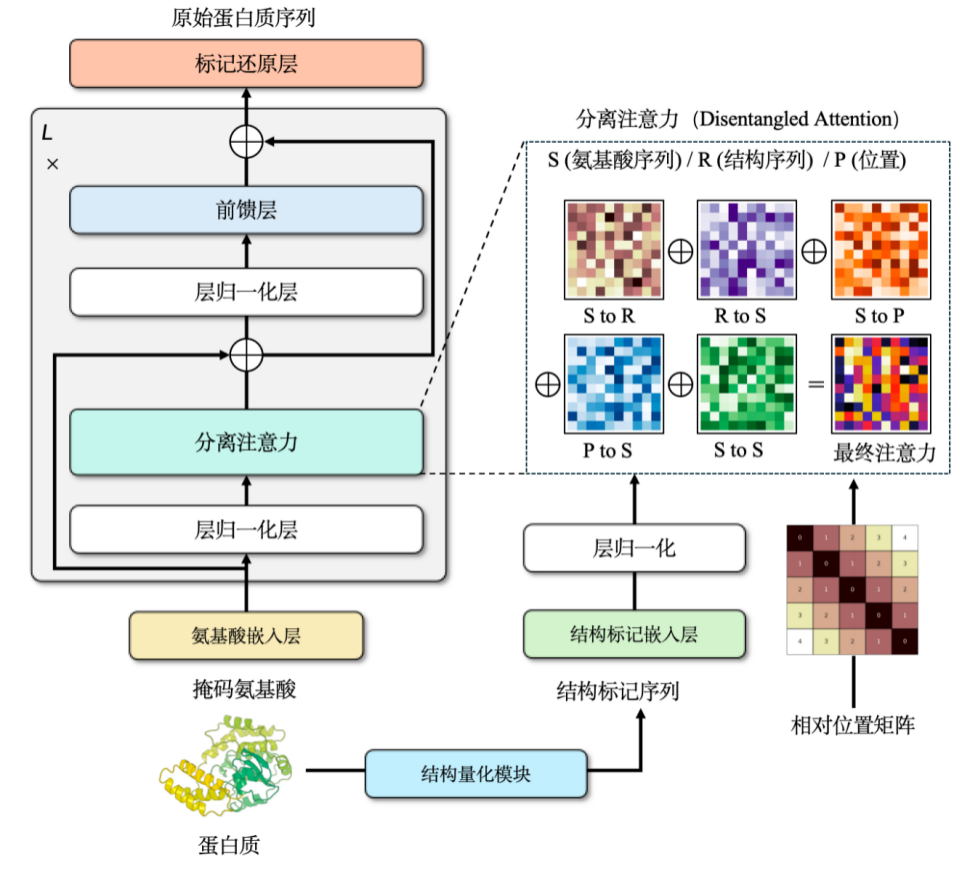
Incorporating structural information into protein language models can significantly improve model performance. However, a problem may arise during this process: if the structure data predicted by AlphaFold is directly used for training, while the loss on the training set gradually decreases, the loss on the validation set or test set gradually increases.The key to solving this problem is to regularize the structural information.In layman's terms, it means simplifying complex data to make it more suitable for model processing.
Protein structures are typically represented as continuous coordinates in three-dimensional space. This needs to be simplified by converting them into discrete sequences of integers. To this end, we used a graph neural network architecture and trained it with a denoising encoder, ultimately constructing a discrete structure vocabulary of approximately 2,048 tokens.
With structural and sequence information,We chose the cross attention mechanism to combine the two.This allows the modified Transformer model to input both amino acid and structural sequences. During the pre-training phase, we designed this model as a language model development task.The training data contains more than 18.8 million high-quality protein structures with a parameter size of approximately 110 million.The model achieved state-of-the-art results at the time, and although it has since been surpassed by newer models, it still held the best results for its class at the time of its publication.
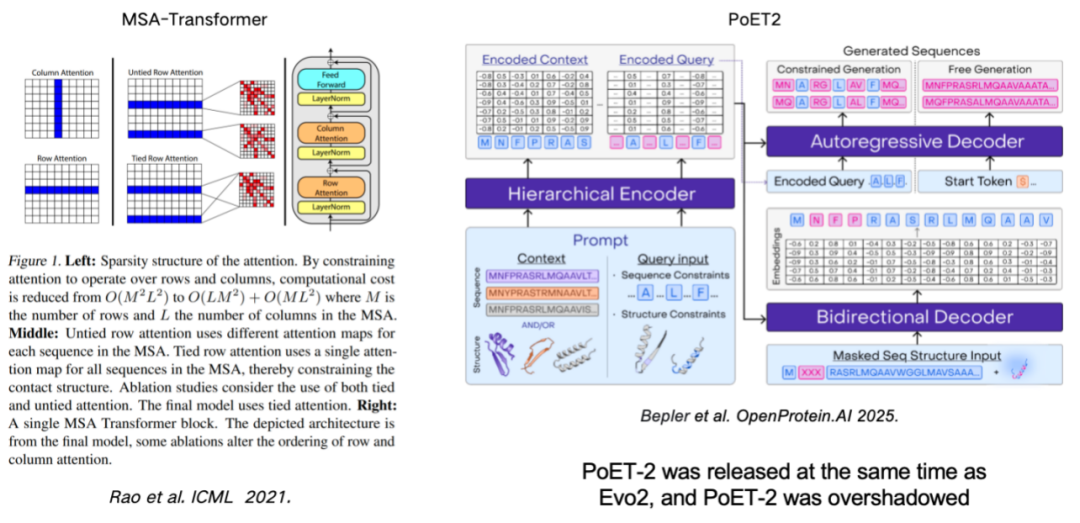
Using MSA (multiple sequence alignment) to enhance the protein language model is also an important means to improve model performance.This work can be traced back to the MSA-Transformer, which effectively incorporated MSA information into the model by introducing row and column rules. The recently released PoET2 model uses a hierarchical encoder to process MSA information and integrates it into a full-pass model architecture. After large-scale training, it has demonstrated outstanding performance.
The Law of Scaling: Is a Bigger Model Always More Powerful?
The so-called Scaling Law originated from the field of natural language processing. It reveals a universal law:Model performance will continue to improve with the increase of parameter scale, training data volume and computing resources.
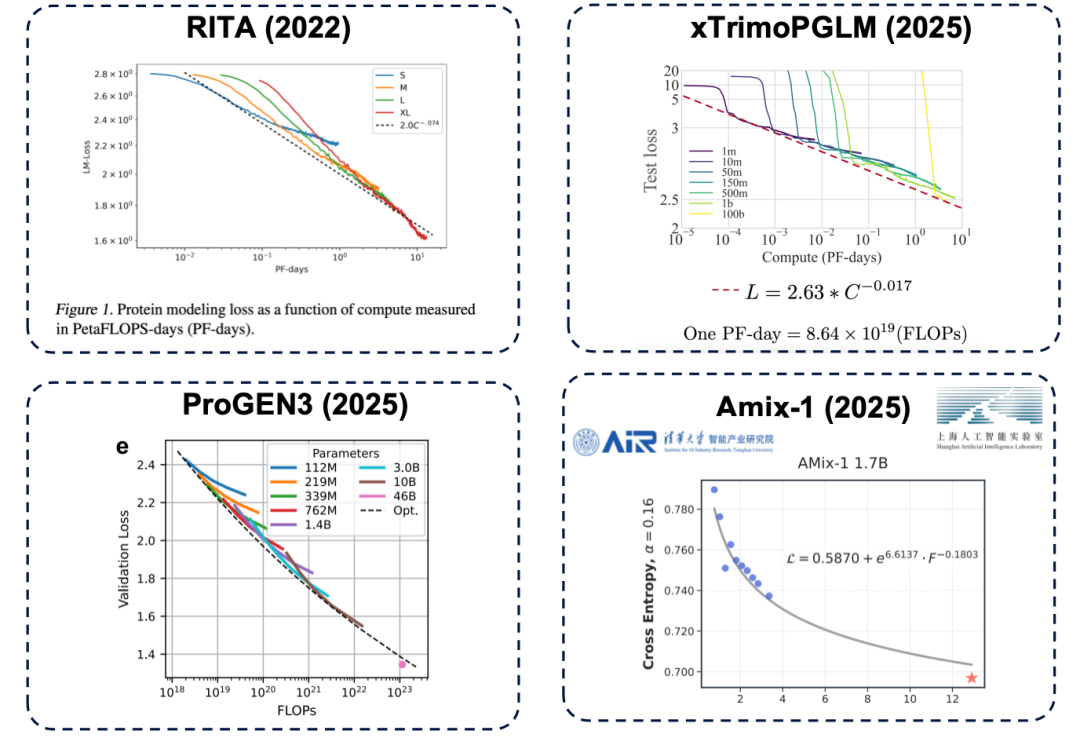
Parameter size is a key factor in determining the upper limit of model performance. If the number of parameters is insufficient, even if more computing resources (in layman's terms, "spending more money") are invested, the model's performance will reach a bottleneck. This same principle also exists in the field of protein language models and has been confirmed by numerous studies, including representative works such as RITA, xTrimoPGLM, ProGEN3, and Amix-1.
* RITA model: developed by Oxford University, Harvard Medical School and LightOn AI.
* xTrimoPGLM model: Developed by the Baitu Bioscience team, it scales the model parameters to approximately 100 billion.
* ProGEN3 model: developed by the Profluent Biotech team.
* Amix-1 model: Proposed by Tsinghua University Institute of Intelligent Industries and Shanghai Artificial Intelligence Laboratory, it uses a Bayesian flow matching network architecture and also has an expansion law.
The "scaling law" we mentioned earlier refers to the pre-training process. However, in protein research, we are ultimately concerned with the performance of downstream tasks. This raises the question:Does improved pre-training performance necessarily help downstream tasks?
In the xTrimoPGLM evaluation, the research team found that in approximately 44% downstream tasks, there is indeed a positive correlation between "better pre-training performance and stronger downstream performance."
At the same time, the Amix-1 model demonstrated emergent capability in structure prediction tasks. This refers to tasks where a small model is completely incapable of solving the problem, but performance suddenly improves significantly when the model's parameter size exceeds a certain critical point. In this experiment, this phenomenon was particularly pronounced in structure prediction tasks, where performance improvement exhibited a "cliff-like red line" when the parameter size exceeded the critical point.
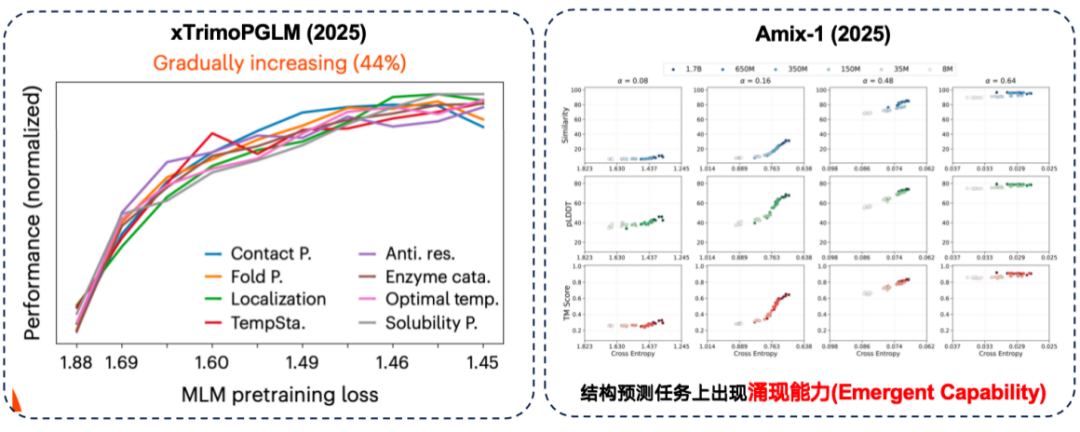
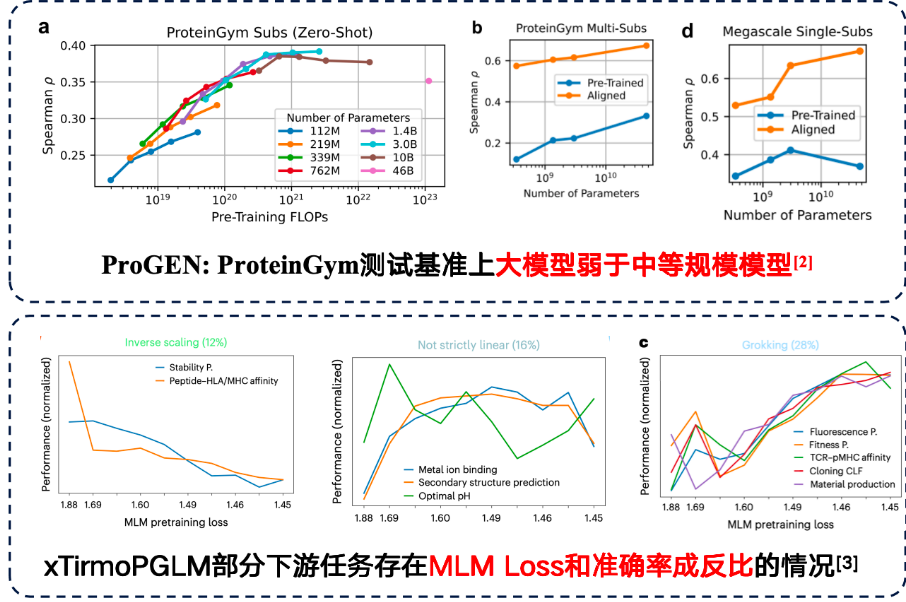
Although in some tasks, large models can indeed bring better downstream performance.However, downstream tasks also discovered an inverse scaling law.That is, the smaller the model, the better the performance becomes.
Studies have shown that simply increasing the number of model parameters does not improve results when the training data itself is highly noisy, so data quality should be given greater attention. In the protein mutation prediction task on the ProteinGym benchmark, medium-sized models actually performed better in terms of accuracy. Furthermore, the team developing xTirmoPGLM also discovered some cases of non-positive correlation, where pre-training performance did not match downstream task performance.
Genomic Modeling: From DNA Design to Protein Yield Optimization
The problem that the genome model solves is: How do we produce a protein?
In synthetic biology,Producing a protein follows the central dogma of molecular biology: "DNA → RNA → protein".In cells, this process is controlled by the cell body, and we can complete this process by designing genes. But the key is that gene design directly affects protein production.
In practical applications, we often encounter situations where a protein has excellent functional performance, but due to poor genetic design, its expression level is extremely low, which cannot meet the needs of industrialization or large-scale application. In this case, AI models can play a role.
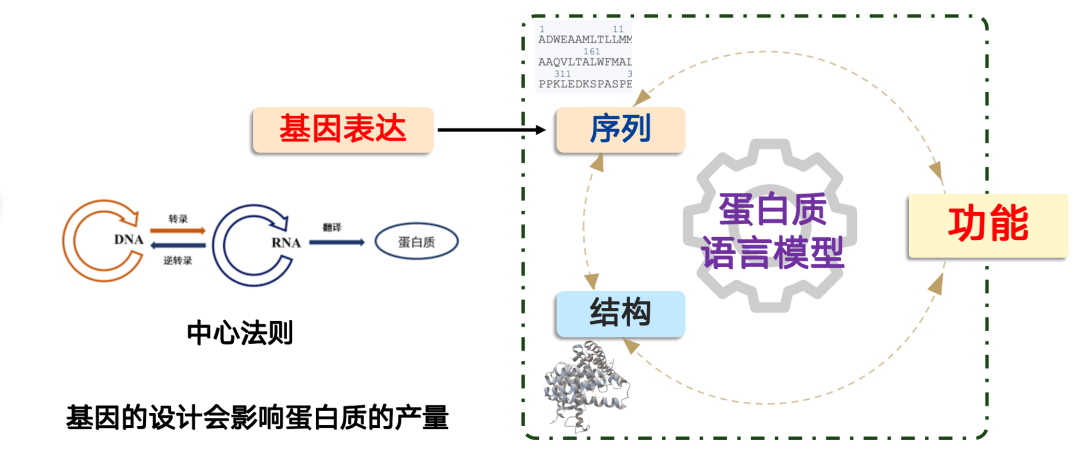
The AI model's mission is to infer how to design DNA sequences directly from protein sequences and increase their production. Our team's proposed model, ProDMM, is based on a pre-training strategy and consists of two phases:
In the first phase, joint pre-training is used to learn representations of proteins and DNA. The inputs include protein and DNA sequences, and a language model is trained using a Transformer architecture. The goal is to simultaneously learn representations of protein, codon, and DNA sequences. In the second phase, generative tasks are trained on downstream tasks, such as from protein to coding sequence (CDS). Given a protein, a DNA sequence can be generated.
* Paper address:Li M, Ren Y, Ye P, et al. Harnessing A Unified Multi-modal Sequence Modeling to unveil Protein-DNA Interdependency[J]. bioRxiv, 2025: 2025.02. 26.640480.
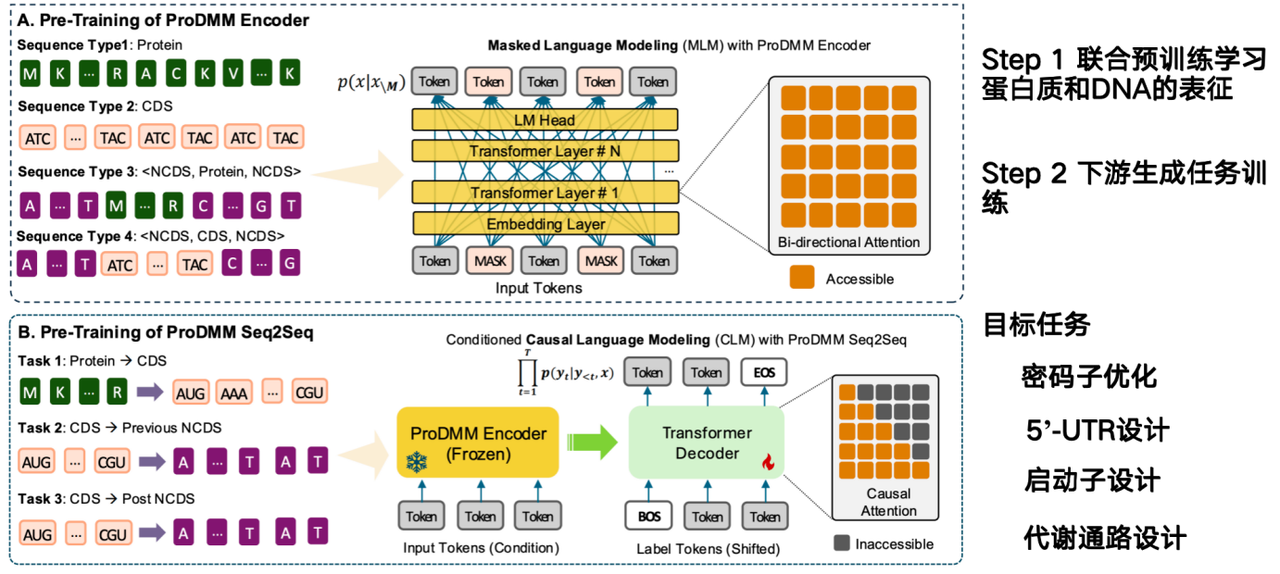
The goal of the project from codons to non-coding DNA (NCDS) is to complete codon optimization, 5'-UTR design, promoter design, and metabolic pathway design.
Metabolic pathway design involves the coordinated work of multiple proteins within a gene to synthesize a specific product. We need to optimize the products of the entire metabolic pathway, a task uniquely suited to genomic models, as protein models optimize only for a single protein and are context-independent. However, a significant challenge facing genomic models is their need to consider the interrelationships within the cellular environment, which is currently their greatest challenge.
About Dr. Li Mingchen
The guest speaker of this sharing session is Li Mingchen, a postdoctoral fellow in Hong Liang's research group at the Institute of Natural Sciences, Shanghai Jiao Tong University. He received a Ph.D. in Engineering in Computer Science and Technology and a Bachelor of Science in Mathematics from East China University of Science and Technology. His main research direction is pre-training protein language models and their fine-tuning.
He has won the title of Shanghai Outstanding Graduate, the National Scholarship, and the Gold Medal of the Shanghai Division of the "Internet+" College Student Innovation and Entrepreneurship Competition. He has published a total of 10 SCI papers as the first author/co-first author/corresponding author in journals and conferences such as NeurIPS, Science Advances, Journal of Cheminformatics, and Physical Review Research, and participated in the publication of 10 SCI papers.
Get high-quality papers and in-depth interpretation articles in the field of AI4S from 2023 to 2024 with one click⬇️









