Command Palette
Search for a command to run...
The Activity of Designed Protein Variants Increased 50-fold! Tsinghua University AIR's Zhou Hao Team Proposed AMix-1 Based on Bayesian Flow Networks to Achieve Scalable and Universal Protein design.

Currently, research in the field of protein pedestal models remains stuck in the "BERT" era, which cannot fully adapt to the biological properties of protein sequences. Previously, AI models such as AlphaFold and ESM significantly advanced developments in multiple fields, including structure prediction, reverse folding, functional property prediction, mutation effect assessment, and protein design.However, these models still lack scalable and systematic methodologies similar to cutting-edge large language models (LLMs), and their capabilities cannot be continuously improved with the increase in data volume, model scale and computing resources.
The lack of universality of such models has brought about difficult-to-solve challenges in the field of protein design: the models cannot capture the conformational heterogeneity of proteins, and predictions about protein design cannot go beyond the scope of the training data; and the excessive reliance on the transfer of NLP methodologies has led to the lack of original architectural designs targeting protein characteristics.
In this context, Zhou Hao's research group at the Institute of Intelligent Industries (AIR) at Tsinghua University, in collaboration with the Shanghai Artificial Intelligence Laboratory, proposed a systematically trained protein base model AMix-1 based on a Bayesian flow network, providing a scalable and general path for protein design.This model adopted the systematic methodology of "Pretraining Scaling Law", "Emergent Ability", "In-Context Learning" and "Test-time Scaling" for the first time, and designed a contextual learning strategy based on multiple sequence alignment (MSA) on this basis, achieving consistency in the general framework of protein design while ensuring the scalability of the model.
The relevant research results were published on the arXiv platform under the title "AMix-1: A Pathway to Test-Time Scalable Protein Foundation Model".
Research highlights:
* A predictable scaling law was established for the protein generation model based on Bayesian flow network;
* The AMix-1 model spontaneously develops a "perceptual understanding" of protein structure through sequence-level training objectives alone, without the need for explicit structural supervision.
* The contextual learning framework based on multiple sequence alignment (MSA) solves the alignment problem in functional optimization, upgrades the model's reasoning and design capabilities in an evolutionary context, and enables AMix-1 to generate new proteins with conserved structure and function;
* Propose a verification cost-guided test-time extension algorithm to enable a new evolution-based design approach when verification budgets increase.
Paper address:
Follow the official account and reply "AMix" to get the complete PDF
More AI frontier papers:
UniRef50 dataset: preprocessing and iterative clustering
The researchers used the pre-processed UniRef50 dataset during model pre-training. This dataset, provided by EvoDiff, is derived from UniProtKB and filtered from UniParc sequences through iterative clustering (UniProtKB+UniParc → UniRef100 → UniRef90 → UniRef50)., containing 41,546,293 training sequences and 82,929 validation sequences. Sequences longer than 1,024 residues were trimmed to 1,024 residues using a random pruning strategy to reduce computational cost and generate diverse subsequences. This iterative process ensures high-quality, non-redundant, and diverse representation of UniRef50 sequences, providing extensive coverage of the protein sequence space for protein language models.
Download the UniRef50 dataset:
Systematic technical solutions
AMix-1 provides a complete set of systematic technical solutions for implementing test-time scaling for protein pedestal models:
* Pretraining Scaling Law:It is clear how to balance parameters, number of samples and computational effort to maximize the model's capabilities;
* Emergent Ability:It shows that as training progresses, the model will emerge with a "perceptual understanding" of protein structure;
* In-Context Learning:It solves the alignment problem in functional optimization, allowing the model to learn reasoning and design in an evolutionary context;
* Test-time Scaling:AMix-1 opens a new approach to evolution-based design as verification budgets increase.
From training and inference to design, AMix-1 has demonstrated its versatility and scalability as a protein foundation model, paving the way for practical implementation.
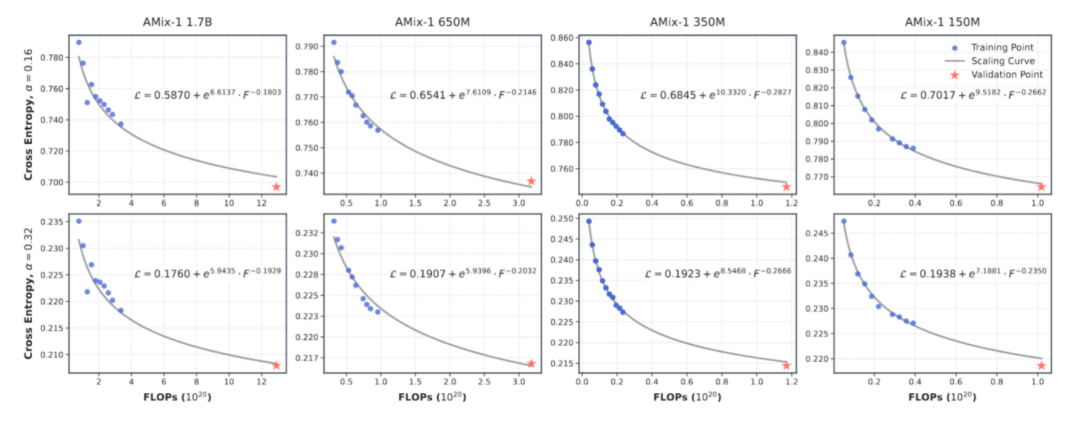
Pretraining Scaling Law: Predictable Protein Model Capabilities
To achieve a predictable scaling law for AMix-1, this study designed a multi-scale model combination with parameters ranging from 8 million to 1.7 billion in the experiment, and used training floating-point operations (FLOPs) as a unified measurement indicator to accurately fit and predict the power-law relationship between the model's cross-entropy loss and the amount of computation.
Judging from the results, the power-law curve between the model loss and the computational effort is highly consistent, confirming that the model training process based on the Bayesian flow network is highly predictable.
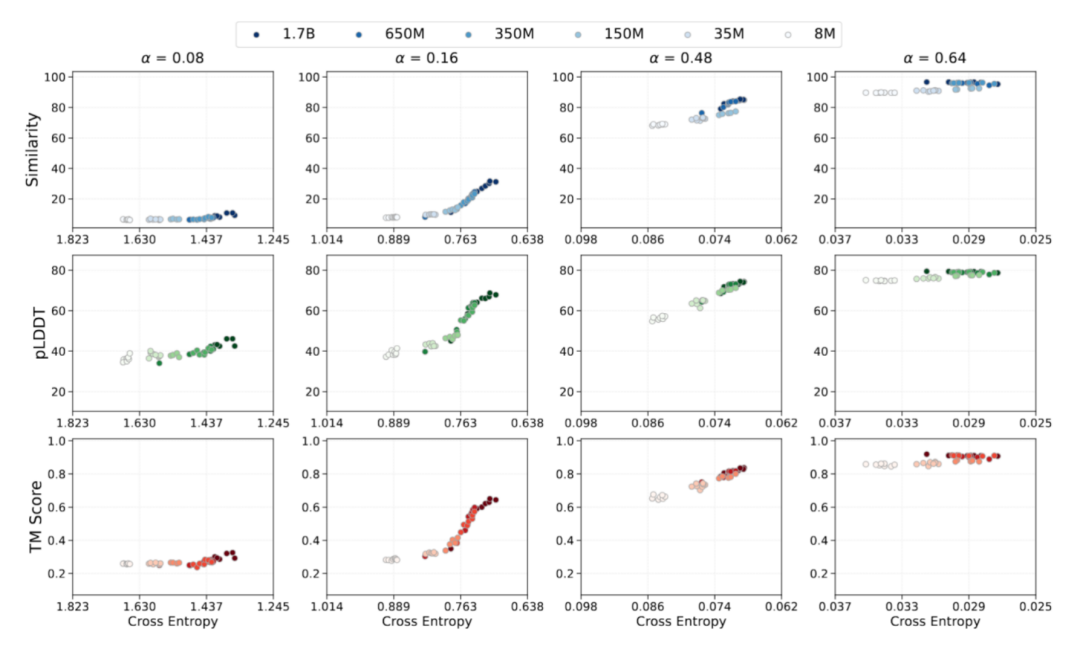
Emergent Ability: Achieving Advanced Model Capabilities
In protein sequence learning, the study of structural emergence is typically based on the "sequence-structure-function" paradigm. To validate the connection between optimization dynamics and functional outcomes in protein modeling, the research team analyzed emergent behavior from a loss-focused perspective based on a predictable scaling law. While using predictive cross-entropy loss as an anchor, they empirically mapped the training loss to protein generation performance. This study's evaluation of the model's emergent ability focused on three aspects:
* The ability of the model to recover sequence levels from corrupted sequence distributions based on sequence consistency observations;
* The transition of models from sequence understanding to structural feasibility from the perspective of foldability;
* Judging the model's ability to maintain structural characteristics from structural consistency.
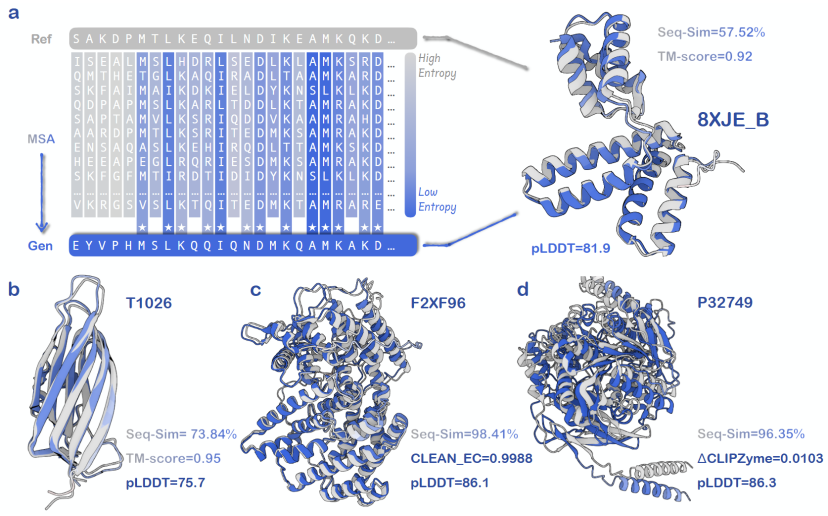
The relevant data during AMix-1 training fully demonstrates the emergence process of the protein base model's "sequence consistency, foldability, and structural consistency" capabilities.The data shows that all the capability indicators of the model during training are highly correlated with the cross-entropy loss, which verifies the possibility of predicting model capability through Scaling Law and cross-entropy loss.At the same time, when trained only with sequence-level self-supervised objectives and without introducing any structural information, the model still exhibits emergency ability after the cross-entropy loss drops to a threshold, showing a nonlinear transition between pLDDT and TM-score.
In-Context Learning: A General Paradigm for Protein Design
Through computer simulation cases, researchers verified the In-Context Learning mechanism of AMix-1. The simulation case experiments showed thatAMix-1 is able to accurately extract and generalize structural or functional constraints from input samples without relying on explicit labels or structural supervision.
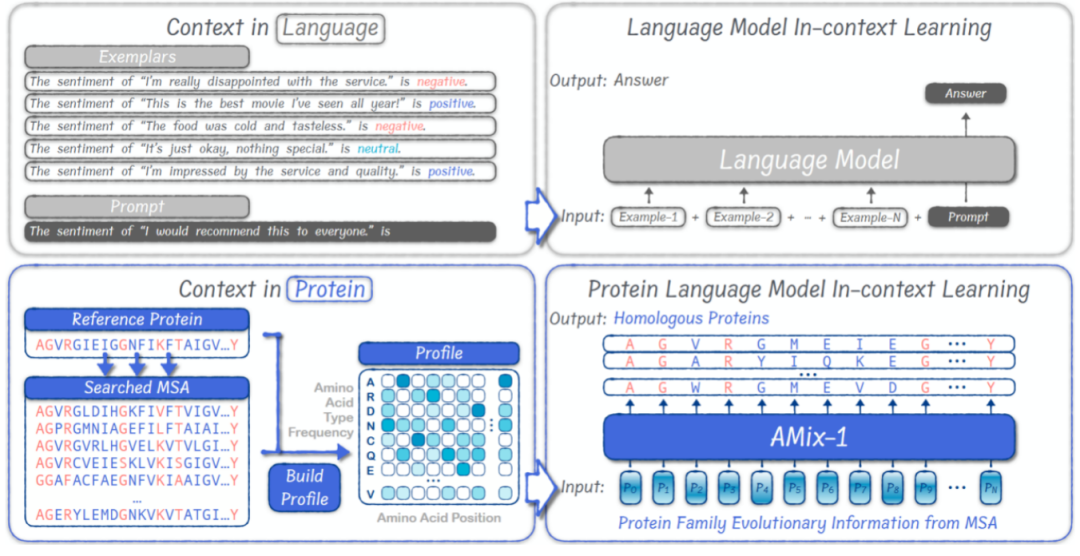
Compared to traditional protein design, which requires customized processes based on task types and lacks a unified protein design framework, AMix-1 introduces an in-context learning (ICL) mechanism within a large language model to accomplish structure- and function-guided protein design. Experiments have shown that, in structural tasks, AMix-1 can generate novel proteins with highly consistent predicted structures using conventional homologous proteins, or even proteins with virtually no homology, as cues. In functional tasks, AMix-1 can generate highly consistent proteases based on the enzymatic function and chemical reaction-guided design of the input protein.
Under this general mechanism,The model can automatically infer the common information and rules in a given group of proteins, and use these rules to guide the generation of new proteins that conform to the common rules.This mechanism compresses a group of protein MSAs into a position-level probability distribution (Profile) input into the model. After quickly analyzing the structure and functional rules of the input proteins, the model can generate new proteins that meet the intention.
Test-time Scaling: Scalable General Intelligence
Based on the test-time scaling approach, researchers employed the Proposer-Verifier Framework to build EvoAMix-1. By continuously increasing the verification budget, they improved AMix-1's model performance. While amplifying the model's design efficiency, the team also achieved scalability. Furthermore, to ensure compatibility, the team eliminated pre-defined requirements for the verifier's properties.
EvoAMix-1 promotes exploration based on the inherent randomness of probabilistic models. By integrating task-specific computer simulation reward functions or experimental detection feedback, it iteratively generates and screens candidate protein sequences under evolutionary constraints. It can achieve efficient directed protein evolution without model fine-tuning, achieving robust and test-time scalable performance in protein design.Across all six design tasks, EvoAMix-1 consistently outperforms AMix-1 in In-Context Learning and various strong baseline methods.
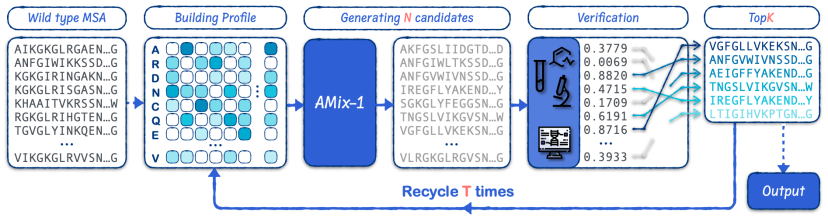
Compared with the traditional method of generating new protein variants through importance sampling,EvoAMix-1 does not update model parameters, but instead builds a proposal distribution through contextual examples.In each round, AMix-1 takes as hints a set of multiple sequence alignments (MSAs) or their spectra, which are considered as input conditions for a protein base model, which then samples neighboring sequences, effectively defining a new conditional proposal distribution.
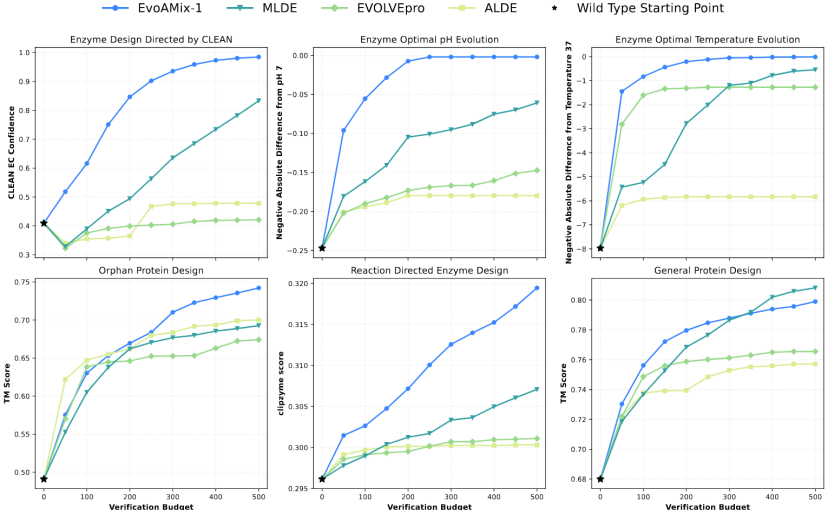
The research team systematically validated the versatility and scalability of EvoAMix-1 across several representative protein directed evolution tasks, including optimal pH and temperature evolution of enzymes, function preservation and enhancement, orphan protein design, and general structure-guided optimization. The experimental results demonstrate the robust scalability of EvoAMix-1 test-time scaling, demonstrating strong versatility across tasks and objectives.
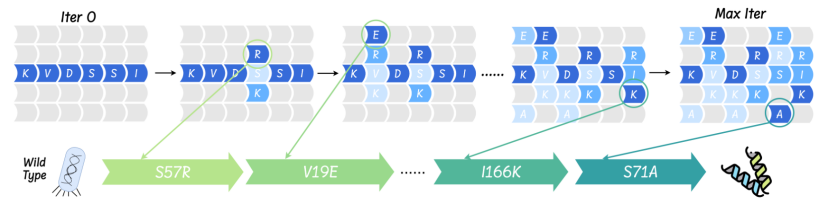
Wet experiment verification: AMix-1 assists in the development of protein AmeR variants with a 50-fold increase in activity
The study tested the "context-cued design" strategy in actual wet experiments, further validating the advantages of AMix-1 in efficiently designing highly active AmeR variants. The researchers selected the target protein AmeR and used the AMix model to generate 40 variants based on the probability distribution of the AmeR family. The inhibitory ability of each variant was evaluated through fluorescent reporter gene experiments. Each variant contained only ≤10 amino acid mutations, and the higher the fold repression value, the stronger the function. In addition, the study proposed a scaling algorithm for evolutionary testing to enhance the applicability of AMix-1 in directed protein evolution, and verified its performance through a variety of computer simulation target area indicators.
The final results show thatThe optimal variant generated by AMix-1 has an activity improvement of up to 50 times, and its performance is improved by approximately 77% compared to the current SOTA model.In addition, AMix-1 does not rely on repeated screening or manual design, but is completely automatically generated by the model.It has achieved a complete closed loop from "model to experiment" and achieved the first-ever breakthrough in AI's use of functional protein design.
Global topology, perception opens a new dimension in protein design
Currently, research on the integration of AI and protein design is booming. In addition to AMix-1, the geometry-aware diffusion model TopoDiff, proposed by Gong Haipeng's team at the School of Life Sciences at Tsinghua University and Xu Chunfu's team at the Beijing Institute of Life Sciences, has also achieved significant breakthroughs in protein design.
Traditional diffusion models such as RFDiffusion not only suffer from coverage bias when generating specific fold types such as immunoglobulins, but also lack quantitative evaluation metrics for the protein's global topology. This study, based on structural databases such as CATH and SCOPe, proposed an unsupervised system, the TopoDiff framework. By learning and leveraging global geometrically aware latent representations, it achieves unconditional and controllable protein generation based on diffusion models. This study proposes a new evaluation metric, "Coverage," which, through a two-stage encoder-diffusion model framework, decouples protein structure into a global geometric blueprint and local atomic coordinate generation, thus overcoming the research challenges of protein fold coverage.
Furthermore, NVIDIA, in collaboration with Mila, the Quebec Institute for Artificial Intelligence in Canada, has overcome the challenge of long-chain prediction using an upgraded all-atom generation model based on the AlphaFold architecture. Traditional methods not only struggle to generate all-atom structures of very long chains (>500 residues), but also fail to explore non-classical folding conformations, such as membrane protein-specific pockets. The research team introduced a probabilistic decision-making mechanism, replacing deterministic folding trajectories with path integral sampling from quantum field theory, thereby increasing the success rate of membrane protein design to 68%.
From geometrically sensing protein folding to designing long chains of 500+ residues, to natural language-driven protein design, to targeting "undruggable" IDPs, AI is expanding the boundaries of protein design capabilities and providing a new paradigm for research in the field. In the future, AI-driven protein design is expected to open up even greater avenues for the development of innovative therapeutics, enzymes, and biomaterials.
Reference Links: