Command Palette
Search for a command to run...
David Baker's Team Proposes a New Approach to Designing Disordered region-binding Proteins, Specifically Targeting Undruggable Targets, in Science.

Since most diseases are directly related to abnormal protein function, proteins play a key role in drug development. When developing new drugs, researchers often use proteins as core drug targets, allowing drugs to bind to partially stable proteins to intervene in the disease process.However, targeting drugs to intrinsically disordered proteins (IDPs) that lack well-defined structural, sequence, and conformational preferences remains challenging.
The traditional method of using antibody targeting is mainly based on the highly specific binding ability of antibodies to specific proteins to achieve the recognition and regulation of target proteins. However, this targeting pathway not only requires a lot of experimental operations, but also disordered antigens are easily degraded and ineffective after injection. Therefore,Proteins with intrinsically disordered regions (IDRs) accounting for more than 50% in the proteome are generally judged as "undruggable" targets and have never been used for drug development.
In this context, David Baker, an outstanding computational biologist who won the 2024 Nobel Prize in Chemistry and director of the Institute for Protein Design at the University of Washington, and his team proposed a protein design strategy called Logos.Based on the induced fit binding strategy, binding proteins that can adapt to 39 target disordered amino acid sequences were designed.This study generated a specialized extended repeat protein backbone and then generalized it using the RFdiffusion model. The backbone incorporates pockets specifically designed for repeat peptide sequences, enabling the designed binder-target peptide template to universally recognize disordered protein regions. This means more proteins can provide targets for new drug development, potentially accelerating research into cancer and Alzheimer's disease.
The relevant research results were published in Science under the title "Design of intrinsically disordered region binding proteins."
Research highlights:
*Establish a template structure library suitable for general recognition to achieve binding adaptation conformation induction for any target sequence.
* Designed binding proteins for 18 synthetic peptide sequences and 21 naturally disordered regions (IDRs) with broad diversity and therapeutic potential, capable of targeting disordered regions of cancer-associated extracellular receptors and driving protein localization within cells.

Paper address:
https://www.science.org/doi/10.1126/science.adr8063
Follow the official account and reply "Natural disordered protein" to get the full PDF
More AI frontier papers:
Template library generation: universal peptide identification
This study combines physical design methods and deep learning design methods as a solution to the IDRs binding problem. Limited by the incompatibility of peptide units with heterogeneous target sequences,The study started with different repeat protein structures, using the diffusion model to reorganize the amino acid binding pockets in different repeat units and differentiate them into different amino acids and conformational templates.This allows for wider recognition of sequences.
To identify peptides in naturally disordered proteins, the study first created a backbone template library. The template library has two characteristics:
*Each template structure should be able to "wrap" the stretched peptide chain conformation and provide a large number of opportunities for interactions such as hydrogen bonding and tight packing, thereby achieving highly specific recognition of the target sequence.
*The template structure is broad and can match any target sequence, so that at least one template can induce it to become a defined and suitable binding conformation.
The process of generating a backbone template library is divided into three steps: backbone generation, protein active pocket specialization, and protein active pocket assembly.
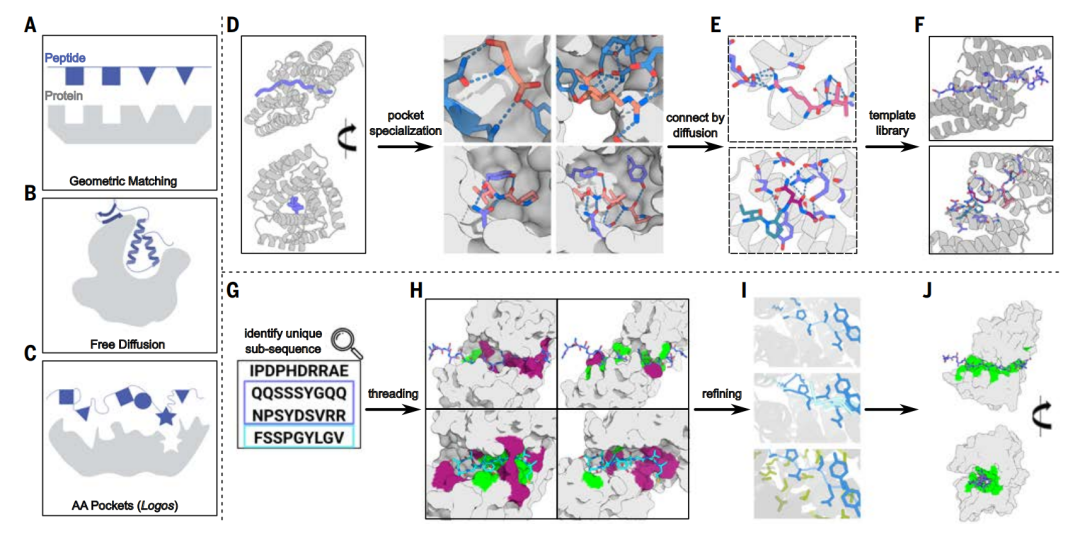
Scaffold Generation
During the backbone generation stage, the research team chose to target multiple extended conformations rather than limiting themselves to the polyproline II conformation, as the polyproline II conformation primarily occurs in proline-rich peptides.
In the stretched conformation, the side chains of the amino acids alternately face opposite directions, which is consistent with the characteristics of a two-residue repeat.The researchers used the Rosetta design method to design a series of dipeptide repeat sequences.Including LK, RT, YD, PV and GA (all are single-letter abbreviations of amino acids), they are designed to entangle and bind with these peptide segments in different stretched conformations, so that each repeating unit interacts with a dipeptide unit.
Subsequently, the researchers characterized these designed four-repeat unit versions of the binding proteins through fluorescence polarization experiments. The results showed that they exhibited nanomolar binding ability for the LK and PV repeat peptides; but had weaker binding ability for the more polar RT and YD, and no binding signal was detected at all for the highly flexible GA.
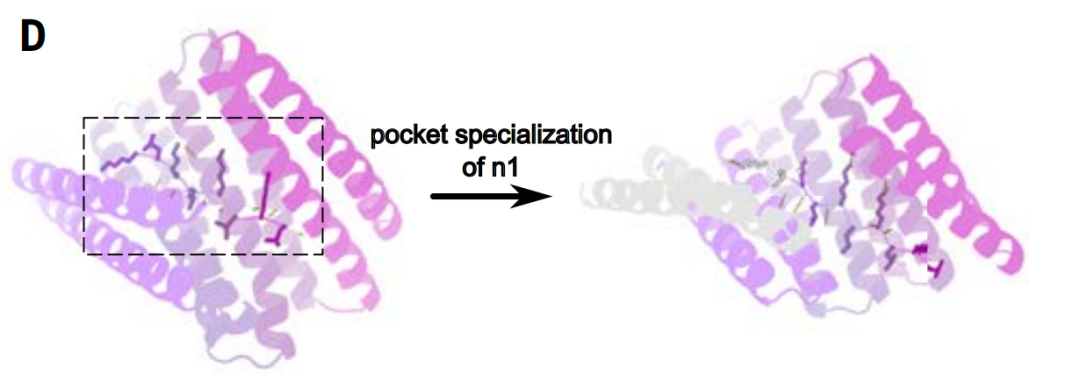
Pocket Specialization
In the step of specifying the protein active pocket, researchers used diffusion modeling to fine-tune the pocket to achieve a more precise match with the specific target peptide sequence.
To improve template matching efficiency, the researchers refined the designed binding pocket, increasing the number of interacting repeat units from four to five while improving the match with the target sequence. This approach also enhanced the affinity between the target structures. The four to nine amino acids surrounding each side-chain bifurcated hydrogen bond between the repeat protein and the peptide backbone were kept fixed, while the hydrophobic interactions between the designed binding proteins were diversified.
The advantage of this strategy is that the geometric configuration requirements of hydrogen bonds are more stringent. In comparison, non-polar hydrophobic stacking has higher spatial freedom. Therefore, in design, it is more efficient to retain hydrogen bonds directly in a template manner rather than repeatedly sampling hydrogen bonds from scratch.
The newly expanded fifth repeat structure is shown in light grey
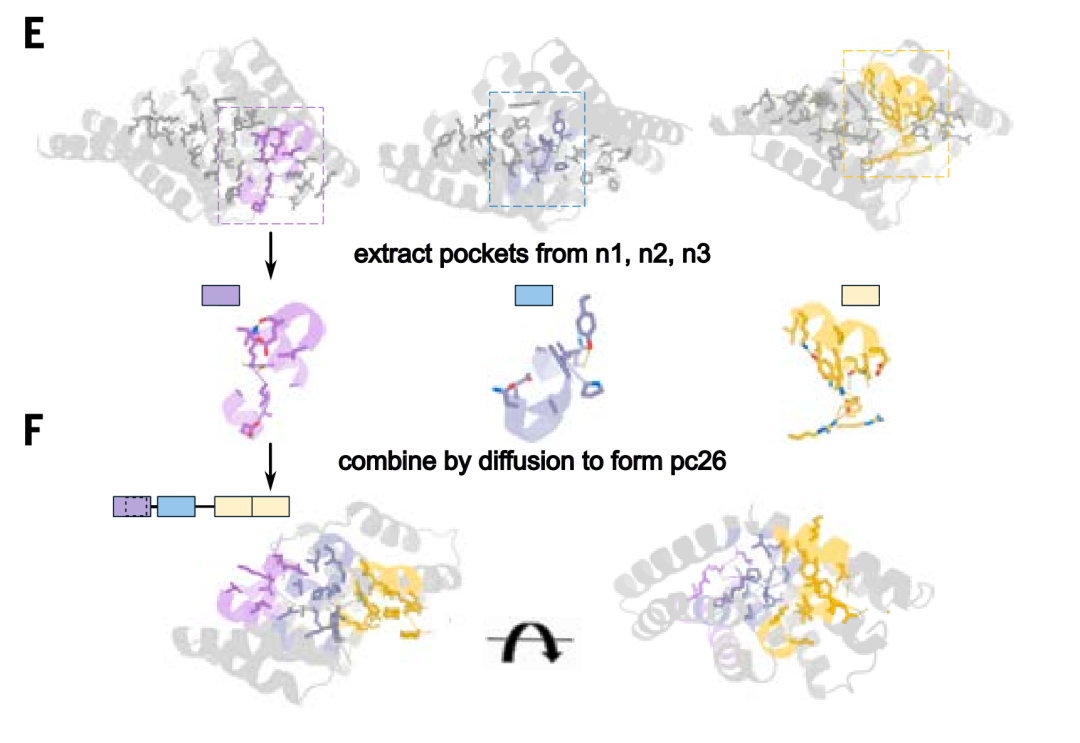
Pocket Assembly
During the pocket assembly step,The researchers used the RFdiffusion model to create interfaces between pockets, resulting in an overall rigid structure and generating a template for assembling the binding pockets into a new backbone.The various pockets in the template are arranged according to different orders and geometries to interact with the peptide target in a series of extended conformations, allowing for more general recognition of non-repetitive sequences.
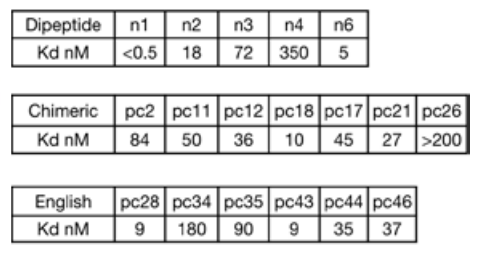
After generating chimeric protein models interacting with chimeric peptide targets, the study parametrically located the binding pockets and connected them via radiofrequency diffusion. Using this approach, the study generated 70 design proposals for seven chimeric targets. Characterization by split-luciferase abundance and biolayer interferometry experiments revealed that, with an average of only 10 designs tested per target, six of the seven targets achieved double-digit nanomolar binding.
To expand the size of the template library to cover a wider range of sequences, the study used pocket assembly technology to construct 36 chimeric backbones containing pockets that recognize polar residues, and generated 1,000 templates consisting of a designed binding protein and a corresponding peptide backbone, in which the amino acids in the peptide conformation can match the designed pockets in the binding protein.
IDR binding protein design and optimization
After establishing a template library, researchers inserted naturally disordered regions into the template library and used it to generate binding proteins that can bind to non-repetitive synthetic sequences and any naturally unstructured target. This step is divided into two parts: threading matching and structural refinement.
Thread matching: determining the most compatible sequence fragment-template pair
In threading matching, the target sequence is threaded into the backbone of each template to identify the most compatible sequence fragment to pair with the template.
Generally speaking, IDP or IDR has a large number of possible peptides that can be used as targets. In order to find the peptide with the greatest targeting potential in IDR,The study first eliminated peptides with low sequence complexity and peptides with multiple close matches in the proteome to prevent cross-reactions with binders of such targets.After local backbone resampling by mapping the unique sequence fragments of the remaining amino acids to the target backbone of the template library,The study used the deep learning-based protein sequence design tool ProteinMPNN to optimize the sequence of the binding protein and evaluated it based on the fit between the designed binding protein and the target sequence and the consistency between the AF2 prediction value and the model.
In cases where AF2 metrics were suboptimal, RFdiffusion was used to customize the backbone for a specific target. Threaded matching was then used to generate binders for therapeutically relevant IDPs, IDRs, and IDP fragments, generating an average of 28 designs per target.
Structural optimization: improving the match between binding protein and target peptide
The best match was also optimized to enhance the fit between the designed binding protein and the target peptide.The study selected the dissociation constant of DYNA_1b1 binding protein and dynorphin for testing, and optimized the radiofrequency diffusion for the highest hit rate of synthetic targets.The results showed that among the 48 designs, 45 showed strong affinity in the screening test, and only the dissociation constants of 6 designs showed weak affinity.
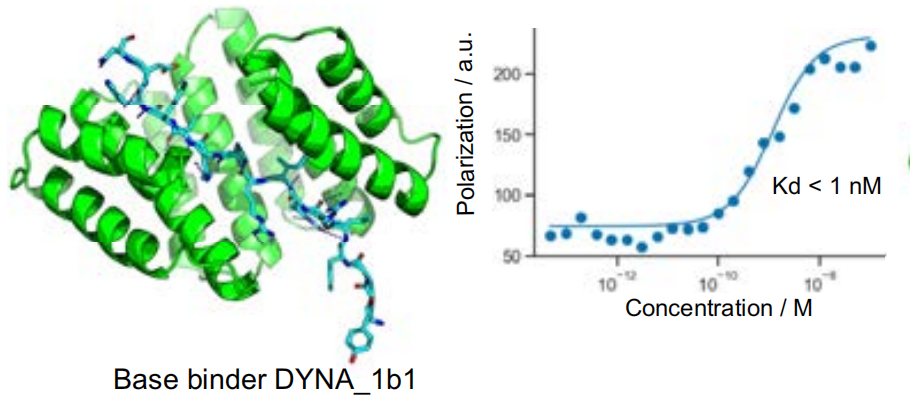
Validation of the orthogonality between dynorphin structure and binding protein
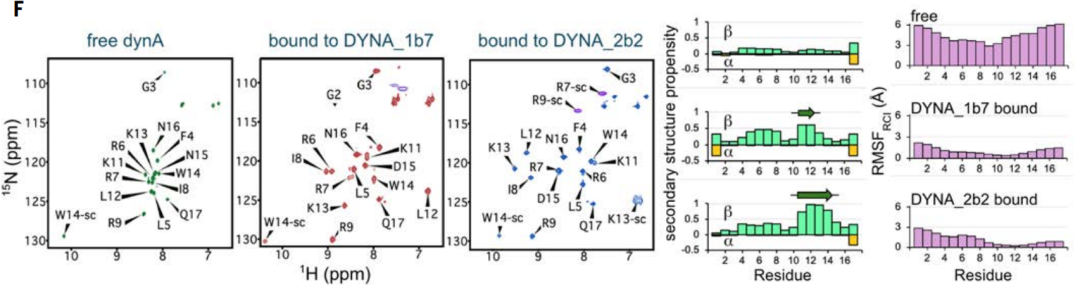
To verify the changes in the dynorphin structure during binding, the nuclear magnetic resonance (NMR) spectra of isotope-labeled dynorphin A were detected in solution when it was unbound, bound to DYNA_1b1, and bound to DYNA_2b2 with higher affinity.
From the NMR results, it was shown that free dynorphin A is intrinsically disordered, but the regions contained in the designed backbone become ordered upon binding.For both bound complexes, NMR data revealed extended bound-state conformations, consistent with the designed model and confirming the effectiveness of dynorphin action in inducing disordered proteins and peptides into non-native conformations.
To explore the optimization potential of Logos, the researchers selected a dynorphin binder, DYNA_1b1, with a binding constant (Kd) of approximately 1 nM. The researchers optimized the top-ranked designs using RFdiffusion. Of the 48 designs, 45 demonstrated strong binding at a concentration of 5 nM via BLI screening, with six having Kd values of ≤100 pM as measured by BLI. Fluorescence polarization measurements of two of the optimized designs (DYNA_2b1 and DYNA_2b2) revealed Kd values below 60 pM and 100 pM, respectively, as shown in Figure B below.
Note: Dynorphin is a kappa-opioid receptor (KOR) peptide ligand associated with chronic pain.
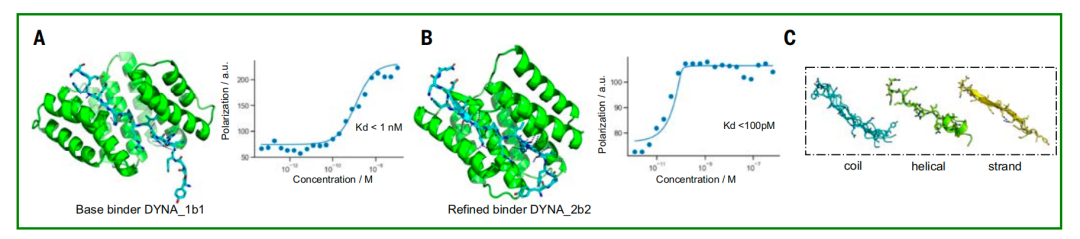
In the original and optimized designs of dynorphin A, the peptide exhibited multiple conformations, including random coils, partial β-strand structures, and partial α-helical structures, as shown in Figure C above. Although dynorphins A and B share a sequence similarity of 62%, their respective binding proteins do not cross each other and only bind to their respective targets. At the same time, the co-crystal structure of the designed protein DYNA_1b7 bound to dynorphin A is highly consistent with the computational design model, especially at the core binding interface (Figures DE above). NMR data also further confirmed thatThe originally disordered dynorphin A's skeleton becomes ordered after binding to the designed protein, once again confirming the effectiveness of the induced fit mechanism (Figure F above).
Verify the functionality and orthogonality of binding proteins
The study used immunoprecipitation models of the WASH complex and the PER complex. The WASH complex includes WASH, FAM21, CCDC53, SWIP, and WASHC2. Testing showed that FAM21_1b1 extracted the entire WASH complex from cell lysates.
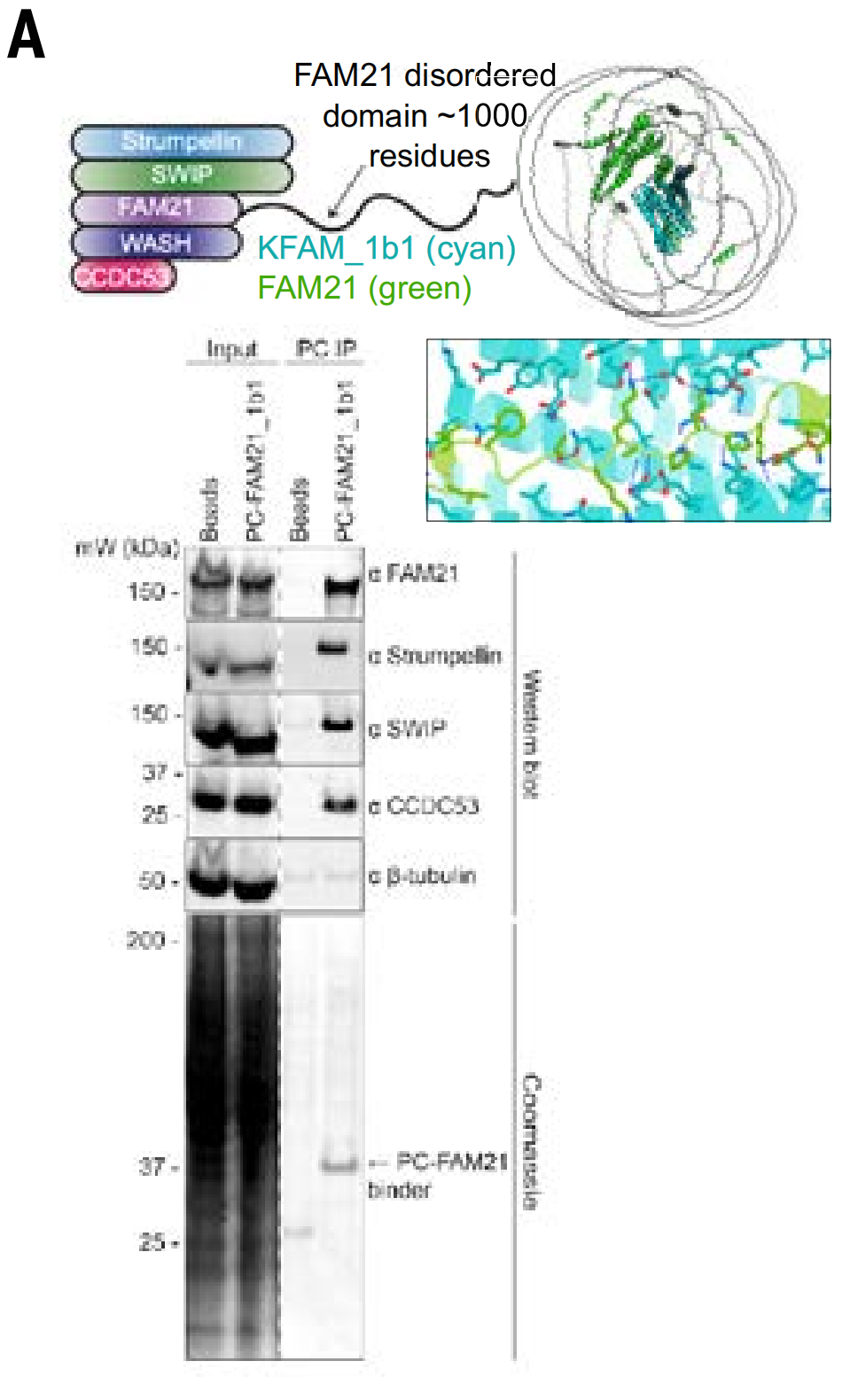
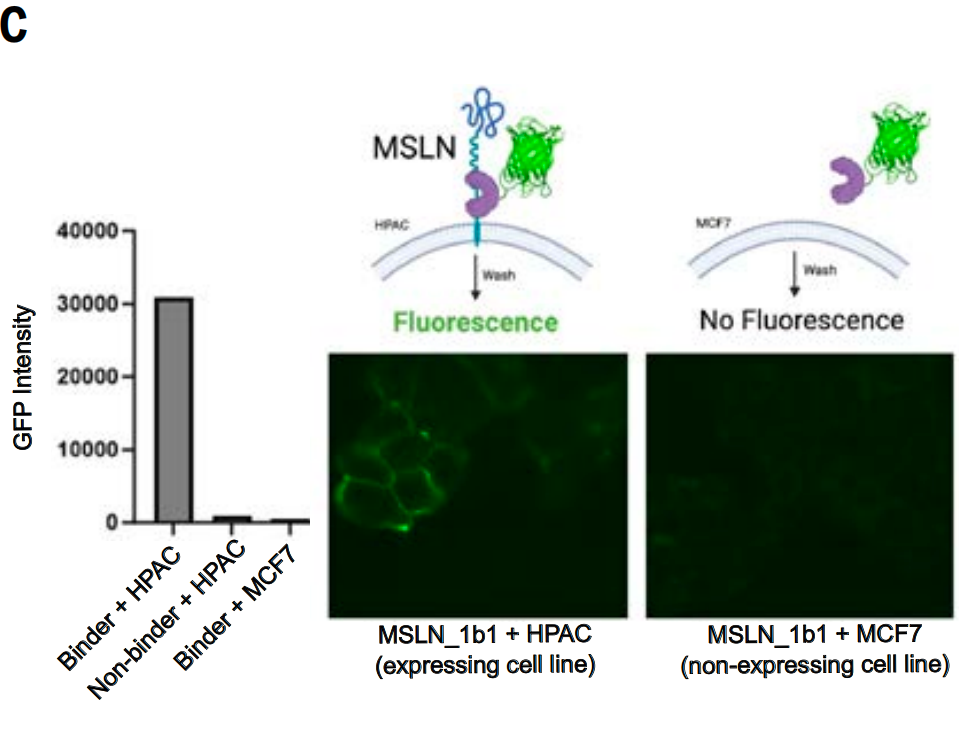
In addition, they also studied whether a binding protein (MSLN_1b1) designed for the MSLN juxtamembrane region can specifically bind to cells expressing this target (because protease cleavage in this region makes the more distal extracellular domain region less suitable as a target).
Note: Mesothelin (MSLN) is a cell surface glycoprotein that is upregulated in many cancers and therefore has attracted much attention in tumor targeted therapy.
The researchers fused green fluorescent protein (GFP) to MSLN_1b1 and incubated it with cells expressing MSLN (human pancreatic adenocarcinoma cell line HPAC) and a cell line that does not express MSLN (Michigan Cancer Foundation breast cancer cell line MCF7), while also including a GFP-fusion protein that does not bind to MSLN as a control.
Fluorescence microscopy revealed that the GFP-MSLN_1b1 fusion protein aggregated at cell junctions in HPAC cells, consistent with the localization characteristics of MSLN. This phenomenon was not observed in MCF7 control cells. Furthermore, the control binding protein showed no binding signal in HPAC cells, as shown in Figure C below. Therefore, MSLN_1b1 specifically recognizes and binds to MSLN on the cell surface.
AI-driven, unlocking new vistas for protein targeting
Currently, AI is increasingly involved in protein-targeted research, propelling research into a new phase of "multi-technique parallelism." In addition to David Baker's team, the George M. Burslem and Ophir Shalem teams at the University of Pennsylvania have also achieved revolutionary breakthroughs in targeted protein research. This team proposed "protein editing" technology, successfully utilizing a split intein system to directly modify the amino acid sequence of proteins after synthesis in living mammalian cells. This is the first time that non-standard amino acids and chemical tags (biotin, fluorophores) have been precisely incorporated into endogenous proteins. The research findings were published in Science under the title "Intracellular protein editing enables incorporation of noncanonical residues in endogenous proteins."
Paper address:
https://www.science.org/doi/10.1126/science.adr5499
Furthermore, a Chinese and international team led by Gao Caixia from the Institute of Genetics and Developmental Biology, Chinese Academy of Sciences, and Li Guotian from Huazhong Agricultural University, drawing on the protein design work of David Baker's team, developed AiCE, a universal protein engineering method based on inverse folding models. Using this AI-driven protein design strategy, they successfully optimized eight protein classes, including deaminases and nucleases, and developed a novel base editor. The research paper, titled "Advancing protein evolution with inverse folding models integrating structural and evolutionary constraints," was published in Cell.
Paper address:
https://www.cell.com/cell/abstract/S0092-8674(25)00680-4
From live cell editing to neuroprotective therapies, from glycosylation innovations to AI-powered multi-chain design, with the continued advancement of AI in biomedicine, teams around the world are using unprecedentedly diverse approaches to tackle the biomedical challenges underlying naturally disordered proteins. The research team's exploration of targeting naturally disordered regions will create new therapeutic approaches for diseases such as cancer and Alzheimer's disease.
Reference Links:








