Command Palette
Search for a command to run...
NVIDIA Achieves Breakthrough in atomic-level Protein Design, Generating Proteins of up to 800 Residues With High Accuracy
It is well known that designing new proteins with specific structures and functions has great application potential in many fields such as drug development and bioengineering. However, it is not easy to achieve this goal, especially in capturing the relationship between protein sequence and structure, which has always been a major challenge in designing proteins from scratch.
In the past, most methods tended to design protein sequence and structure separately.For example, first generate the sequence and then fold it, or first design the backbone and then determine the sequence. However, it is still a very challenging problem to accurately model the joint distribution of protein sequence and all-atom structure to achieve fine control of functional sites and complete key protein design tasks such as atomic motif scaffold design. This requires not only dealing with discrete sequences and continuous coordinates, but also dealing with the problem of side chain dimensions changing with sequence.
In this context,NVIDIA's research team and Mila, the Quebec Institute for Artificial Intelligence in Canada, proposed La-Proteina.This is an atomic-level protein design method based on partial potential flow matching. It can effectively combine explicit backbone modeling and fixed-size potential representation of each residue to capture sequence and atomic side chain information, solving the key challenge of dimensional variability of explicit side chain representation in protein generation, and bringing new breakthroughs to the field of protein design.
The relevant research results were published on arXiv under the title "La-Proteina: Atomistic Protein Generation via Partially Latent Flow Matching".
Research highlights:
* A partially implicit flow matching framework La-Proteina is proposed, which is designed for the joint generation of protein sequences and complete atomic-level structures. It effectively combines explicit main-chain modeling and a fixed-size implicit representation of each residue to capture both sequence and atomic-level side chains.
* In extensive benchmark experiments, La-Proteina achieves SOTA performance in unconditional protein generation, capable of generating diverse, co-designable, and structurally valid fully atomic-scale proteins of up to 800 residues.
* The study successfully applied La-Proteina to indexed and non-indexed atomic-level motif scaffold design, which are two important conditional protein design tasks, and both demonstrated that the model is superior to previous all-atom generators.

Paper address:
More AI frontier papers:
https://go.hyper.ai/owxf6
Dataset: used to train unconditional models, as well as protein data features and functions
This study used 2 datasets to train unconditional models:
One is the AFDB dataset clustered by Foldseek, which is derived from the screening and clustering of the AlphaFold database (AFDB).The clustering combines sequence and structural information, with an initial set of approximately 3 million unique samples, which were optimized using multiple criteria: an average pLDDT score of no less than 80, a protein length ranging from 32 to 512 residues, a coiling ratio less than 50%, and no more than 20 consecutive coiling residues. The presence of β-folds was specifically required to correct for the low β-fold content in the model-generated proteins.Finally, about 550,000 protein samples were obtained.This dataset has been carefully screened to make the proteins generated by the model more balanced in structural features, especially improving the β-fold content.
The second is a customized AFDB subset for long sequence training.The researchers screened samples from AFDB with an average pLDDT of at least 70 and a length between 384 and 896.After clustering, more than 4 million clusters were obtained for training.Focusing on longer protein samples, it meets the needs of long sequence training.
In addition, the protein data itself contains sequence (20 residue types) and 3D structure information, which are uniformly stored with the help of the Atom37 representation. The Atom37 representation defines a standardized superset of 37 potential atoms for each residue. A protein structure of L residues can be stored as a tensor of shape [L, 37, 3], and the relevant coordinate subsets are selected according to the type of each residue.
The characteristic of this standardization method is that it provides a unified storage and representation method for the structural information of different residues, which lays the foundation for the model to uniformly process the structural information of different residues. The large-scale data characteristics of AFDB provide rich samples for the model, which helps it learn a wider range of protein sequences and structural features, improve performance and generalization capabilities. Through training and experiments with these data, related models can better capture the relationship between protein sequence and structure and achieve more precise design.
La-Proteina: Innovative architecture and training mechanism of atomic-level protein design model
La-Proteina is an innovative model for atomic-level protein design. Its core design revolves around "partial implicit representation" and aims to solve the complex challenges in generating fully atomic-level structures.
At the design level, considering the challenge of generating all-atom structures while taking into account the large-scale backbone, amino acid types, and side chains (the side chain dimensions vary with amino acids),La-Proteina proposes to encode the atomic-level details and residue type of each residue into a continuous latent space of fixed length while maintaining explicit backbone modeling through α-carbon coordinates.
This design brings multiple advantages - it not only avoids the difficulty of mixed continuous-classification modeling in the main generation component of the model, allowing the full continuous flow matching method to efficiently generate hidden variables, but also can be based on the advancement of high-performance main-chain modeling. At the same time, explicit main-chain modeling allows different generation schedules to be set for the global α-carbon backbone and residue atomic-level details, which is the key to high performance and also improves scalability, enabling the model to be expanded to large proteins of up to 800 residues. This hybrid approach is the core reason why it is superior to the fully implicit modeling framework.
From the composition structure, as shown in the figure below,The core of La-Proteina consists of three neural networks: encoder, decoder and denoiser.All three share the core Transformer architecture based on the biased attention mechanism.
Among them, the encoder is responsible for mapping the input protein (containing sequence and structure information) to latent variables. Its initial sequence representation covers the original atomic coordinates, side chain and backbone torsion angles and residue types, and the initial pair representation includes the relative sequence separation, pairwise distance and relative direction between residues; the decoder is responsible for reconstructing the complete protein from the latent variables and α-carbon atom coordinates, processing 8-dimensional latent variables and α-carbon atom coordinates for each residue; the denoiser network is used to predict the velocity field that transfers samples from the standard Gaussian reference distribution to the target data distribution, and directly conditions the interpolation time in its Transformer block.
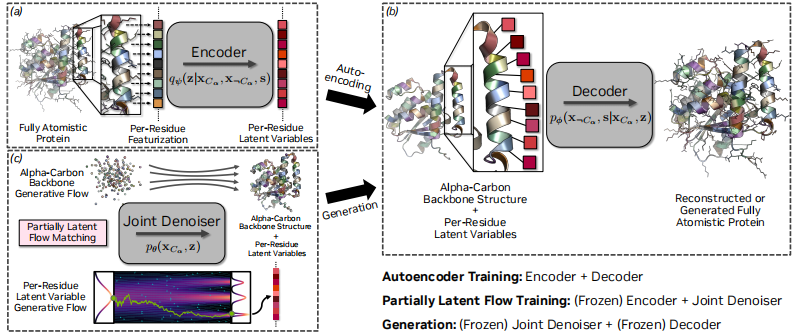
In terms of training methods,La-Proteina uses a two-phase training strategy.
The first stage trains a conditional variational autoencoder (VAE): the encoder maps the input protein to latent variables, and the decoder reconstructs the protein based on the latent variables and the α-carbon atomic coordinates. The entire VAE is optimized by maximizing the β-weighted evidence lower bound (ELBO). For the above modeling choices, the reconstruction term can be simplified to the cross-entropy loss of the sequence and the squared L2 loss of the structure.
The second stage optimizes the flow matching model to approximate the target distribution. The denoiser network is trained by minimizing the conditional flow matching (CFM) objective. The use of two separate interpolation times tx and tz is the key design of this stage. This setting enables different integration schedules for the α-carbon atomic coordinates and latent variables during inference, effectively enhancing model performance.
Through such design and training, La-Proteina is able to efficiently learn the joint distribution of protein sequences and all-atom structures, providing strong technical support for atomic-level protein design.
Experimental results: La-Proteina leads by a large margin in all four tests
To verify the performance of La-Proteina, the research team conducted a series of experiments around two major directions: unconditional atomic-level protein generation and atomic motif scaffold design, comprehensively considering the performance of the model in different scenarios.
In the unconditional atomic-scale protein production experiment,As shown in the figure below, the research team compared two variants of La-Proteina (with and without triangular multiplication layers) with multiple publicly available all-atom generation baseline methods such as P (all-atom), APM, and PLAID. The evaluation indicators covered all-atom collaborative design capabilities, diversity, novelty, and standard design capabilities.
The results show that the two variants of La-Proteina outperform all baseline methods in terms of all-atom co-design capability, design capacity and diversity, and are also highly competitive in terms of novelty.
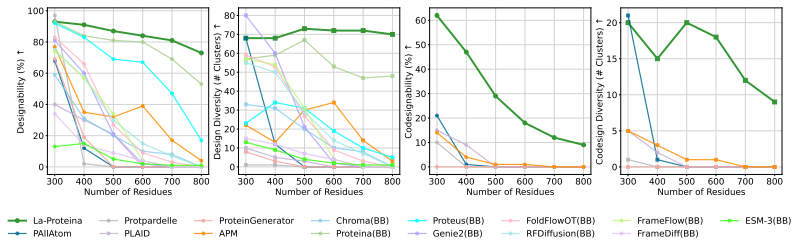
La-Proteina's ability to generate unconditional long chains
It is worth noting that La-Proteina, which does not use triangular multiplication layers, achieves state-of-the-art performance while having high scalability, while the second-best performing P (all-atom) can only process short proteins due to its reliance on computationally expensive triangular update layers.
also,The research team also demonstrated the scalability of La-Proteina in generating large all-atom structures.Trained on the AFDB dataset containing approximately 46 million samples, the model performs best in the task of generating proteins with a length of more than 500 residues, while other all-atom baseline methods often have difficulty generating effective samples in this length range.
In biophysical analysis, the MolProbity tool was used to evaluate the construct validity.The results showed that the structure generated by La-Proteina was of higher quality.The score is significantly better than all baseline methods, and the generated structure is more realistic at the physical level and more similar to the real protein; at the same time, by visualizing the distribution of side chain dihedral angles and comparing them with PDB and AFDB references, it is found that, La-Proteina can accurately simulate the conformational space of amino acid rotational isomers,The baseline methods often deviate from the reference, missing patterns or filling in unrealistic angle regions.
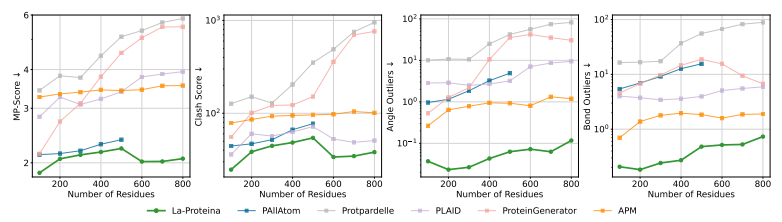
La-Proteina is better than the existing all-atom generation baseline
Has higher construct validity
In the atomic motif scaffold design experiment,The research team evaluated the model's performance on the atomic motif scaffold design task, which requires the model to generate a protein structure that can accurately support the motif based on the atomic structure of a predefined motif. The experiments were conducted in four evaluation settings, including all-atom and advanced atomic scaffold design, as well as indexed and non-indexed versions.
The results show that in all four settings,La-Proteina significantly outperforms the only comparable all-atom baseline method, Protpardelle, and is able to successfully solve most benchmark tasks.Especially for motifs consisting of 3 or more different residue segments, the non-indexed version of La-Proteina performs better than the indexed version, probably because fixing the positions of multiple segments limits the flexibility of the model to explore different structural solutions.
Scientific breakthroughs and innovative practices in the field of atomic-level protein design
In the field of protein design, the atomic-level protein design research direction represented by La-Proteina has attracted widespread attention from academia and the business community. Many universities and companies have achieved important scientific breakthroughs and innovative practices in this field.
In academia, some research teams are working to improve the performance and scalability of protein generation models. For example, NVIDIA has teamed up with Mila, the Quebec Institute for Artificial Intelligence, the University of Montreal, and the Massachusetts Institute of Technology toThe developed Proteina was trained on the large-scale AlphaFold database (AFDB).Demonstrated the scalability of a flow-based model for protein structure generation.
There are also some studies that use diffusion models in protein design. For example, early diffusion-based protein generators such as RFDiffusion and Chroma focus on backbone generation. Subsequent studies have further expanded the scope of application of diffusion models in protein design, such as diffusion on SO(3) manifolds and Euclidean flow matching methods.
Some research teams also focus on the joint modeling of protein sequence and structure. For example, ProtComposer launched by NVIDIA and MIT uses auxiliary statistical models and 3D primitives to generate protein structures, while some works deal with all-atom structures by jointly modeling protein backbone and sequence or using latent variable models. In addition, language models have also been applied to protein design, with some methods focusing on protein sequences, while others tokenize structural information and jointly model sequences and structures.
In the business world, Cradle, a Dutch biotechnology company, focuses on using artificial intelligence to simplify the protein design process. It has established a wet lab to accumulate billions of protein sequences and data to train proprietary generative artificial intelligence models, making protein design and optimization more convenient. Xaira Therapeutics, an American AI pharmaceutical service provider, is committed to creating adaptive molecules for specific indications with its advantages in advanced machine learning research, large-scale data generation, and therapy development. Some companies are also committed to combining protein design technology with artificial intelligence and machine learning to improve the efficiency and accuracy of protein design.
The scientific research breakthroughs of these universities and the innovative practices of enterprises have provided rich experience and technical support for the development of protein design, and promoted the continuous development of this field. With the continuous advancement of technology, it is believed that protein design will play an important role in more fields in the future.
Reference articles:
1.https://mp.weixin.qq.com/s/7r69S3XpNMjemo3EiXzNeQ
2.https://mp.weixin.qq.com/s/DrZEdsb1SqSSkv_hbrp3TA








