Command Palette
Search for a command to run...
Simultaneously Processing Protein Main Chain and Side Chain Information, Stanford Et al. Achieved full-atom Structure Modeling Based on Message Passing Neural Network
Protein sidechain conformation refers to the specific spatial arrangement of the side chains of amino acid residues in proteins in three-dimensional space. Studying protein sidechain conformation can help people understand the relationship between protein structure and function, and has great application value in protein engineering, drug design and other fields. However, current protein sequence design methods based on deep learning mainly focus on fixed main chain protein sequence design, and most of them are unable to model protein sidechain conformation during sequence generation.The key side chain interactions are inferred only based on the main chain geometry and known amino acid sequence tags, while ignoring the role of protein side chain conformation in proteins.
To fill this gap, a team from Stanford University and the Arc Institute in Palo Alto, California,Together, we proposed a new protein sequence design method, FAMPNN (Full-Atom MPNN), which can explicitly model the sequence identity and side chain structure of each amino acid residue.The model uses a message passing architecture based on graph neural networks (GNNs), combined with improved MPNN (Message Passing Neural Networks) and GVP (Geometric Vector Perceptron) modules for all-atom encoding, and can simultaneously process protein main chain and side chain information. Studies have shown that FAMPNN can significantly improve the quality of protein sequence design and the accuracy of experimental predictions by explicitly modeling all-atom structures.
The research result, titled "Sidechain conditioning and modeling for full-atom protein sequence design with FAMPNN", was selected for ICML 2025.
Research highlights:
* We introduce a method that combines cross-entropy and diffusion loss objectives to model the per-label distribution of both the discrete sequence identity of residues and the continuous side-chain structure.
* We implement a lightweight iterative sampling method for generating samples from a joint distribution and use improved MPNN and GVP layers for all-atom encoding
* Research has shown that FAMPNN can effectively improve the accuracy of sequence design and experimental protein fitness prediction by explicitly modeling all-atom structures

Paper address:
More AI frontier papers:
https://go.hyper.ai/owxf6
Datasets: Diverse datasets optimize model training and evaluation
To ensure the effectiveness and reliability of the model, the research team used complex multi-datasets for training and evaluation:
The study mainly used the S40 dataset of CATH 4.2.The dataset is a curated set of domains extracted from the Protein Data Bank (PDB), with redundant domains with homology exceeding 40% removed, and divided into training, validation, and test sets in a ratio of 8:1:1.
The PDB dataset is built based on the entire PDB database and includes structures published as of September 30, 2021. The researchers clustered the proteins according to the sequence homology of 40% at the protein chain level, and prioritized multi-chain protein examples for training models to learn to design multi-chain proteins.
The CASP13, 14, and 15 datasets are mainly used to evaluate the model's performance in side chain packing.The research team used MMseqs2 suggestion search to remove all sequences with homology greater than 40% with the CASP13, 14, and 15 datasets from the training and validation datasets, and then measured the side chain packing performance by the average root mean square deviation (RMSD) between the predicted side chains and the true side chains.
The SKEMPlv2 dataset was used to evaluate the model’s predictive ability for protein–protein binding affinity.The dataset collates the experimentally measured binding affinities of thousands of sequence variants in hundreds of protein-protein interactions, and after processing, the final dataset is 6,649 data points.
The S669, Megascale, and FireProtDB datasets were used to evaluate the model’s zero-shot prediction ability for protein stability.These datasets contain experimental measurements of stability changes of a variety of natural proteins (△△G), which are widely used benchmark datasets for stability predictors. The Megascale dataset is a selected, deduplicated version of the dataset. The research team merged the training set, validation set, and test set into a single dataset, ultimately obtaining a dataset containing 272,712 experimental data points involving 298 different proteins. The FireProtDB dataset contains free energy changes of 3,438 single mutations of 100 unique proteins, of which 3,420 examples were ultimately used after processing. The S669 dataset contains experimental measurements of 669 single mutations of 94 proteins, and 4 variants were excluded from the dataset due to the presence of non-standard amino acids.
The CR9114, CR6261, and G6 datasets were used to evaluate the performance of the model in predicting antibody-antigen binding affinity.Among them, the CR9114 dataset contains all possible combinations of 16 amino acid substitutions. The CR6261 dataset contains all possible combinations of 11 amino acid substitutions, with a total of 65,536 and 2,048 sequences, respectively. The G6 dataset has a total of 4,275 data points binding to VEGF-A.
Intelligent tool to understand protein sequence and side chain structure at the same time
The core goal of this study is to allow the model to learn protein sequence and side chain conformation simultaneously. To this end, the research team used Masked Language Modeling to train FAMPNN based on sequence consistency.The training is done in an end-to-end manner, combining categorical cross entropy loss (for sequence prediction) and diffusion loss (for side chain conformation prediction).This enables the model to simultaneously restore the masked sequence and its corresponding side chain conformations based on partially known sequences and side chain coordinates.
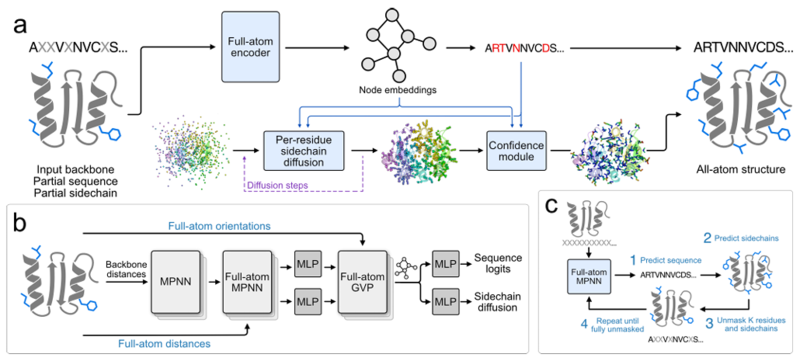
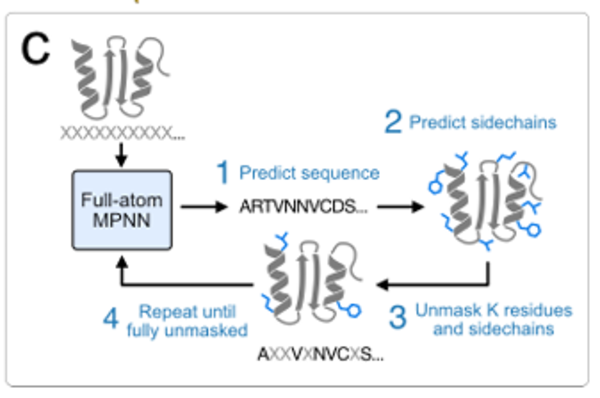
In terms of sampling, an iterative sampling strategy similar to MaskGIT is introduced, starting from the state where the sequence and side chain are completely masked, and gradually predicting and removing the mask of some sequences and side chain tokens until the complete sequence and side chain structure is obtained. As shown in the figure below:
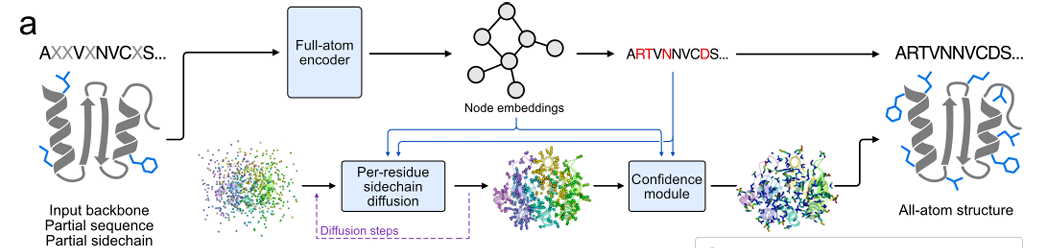
In the specific design, the side chain coordinates are represented in atom37 format.Each residue is a 37 x 3 fixed-size matrix with 37 atoms including the 3D coordinates of 4 main chain atoms (N, Cα, C and O) and 33 side chain atoms. For side chains where no specific atom type exists, ghost atoms (set to the Cα position of the residue) are used to represent them. This method solves the problem of different number of side chain atoms for different amino acids.
As the core of feature extraction, the all-atom encoder uses a hybrid MPNN-GVP architecture graph neural network for encoding.The architecture consists of three main components: the invariant main chain encoder, the invariant all-atom encoder, and the equivariant all-atom encoder. The first two components are built on the architecture of ProteinMPNN. The invariant main chain encoder is the same as the MPNN encoder, which only encodes the main chain structure; the invariant all-atom encoder replaces the MPNN decoder, which is the same as the main chain encoder MPNN encoder, but the characterization is extended to all atoms. The last component uses an improved GVP to enable the model to reason about vector-valued interatomic orientations in addition to the previously encoded scalar-valued interatomic distances.
In terms of side chain coordinate generation, the research team adopted the per-token Euclidean Diffusion method.The core of this method is to use the Euclidean diffusion model (EDM) to solve the problem of generating continuous values of side chain atomic coordinates. The goal is to generate a side chain structure that matches the main chain structure and the spatial arrangement of surrounding amino acids. During training, random noise is first added to the real side chain coordinates, and then the model is allowed to remove the noise and restore the real coordinates based on the noise level and known information; during inference, starting from the random noise coordinates, the model gradually removes the noise and generates side chain coordinates that are close to the real ones.
At the same time, in order to avoid the influence of the overall rotation and translation of the protein on the side chain generation, the side chain atomic coordinates are converted to a local coordinate system based on the main chain atoms during training, and then converted back to the global coordinate system after generation.The input of the diffusion model includes the features extracted by the all-atom encoder, the predicted sequence identity and the current noise level.The generated side chain coordinates are also used in the joint loss function to guide model training, as shown in the figure below.
In order to reduce the error of model prediction and improve accuracy, the research team also designed a confidence module for predicting sidechain packing error (Predicted Sidechain Error, pSCE). Specifically,This module divides the actual error of side chain atoms (the distance between the generated coordinates and the true coordinates) into 33 intervals.The model is trained with categorical cross entropy loss, so that it can predict the interval of each atomic error based on the information in the generation process, and then obtain the final error estimate pSCE through the expectation of the interval probability. The input of this module includes the characteristics of the all-atom encoder, the generated sequence and side chain coordinates, and the noise level in the diffusion process. The output pSCE can effectively reflect the accuracy of side chain packing, which is helpful for screening high-quality design results and enhancing the interpretability of the model, thereby improving the quality assessment link of side chain structure generation.
Experimental results: The performance is significantly better than the model based only on the main chain
To verify the performance and accurately evaluate the model, the research team first conducted sequence recovery and self-consistency evaluation experiments, and benchmarked FAMPNN against other methods. The specific comparison objects are shown in the figure below.
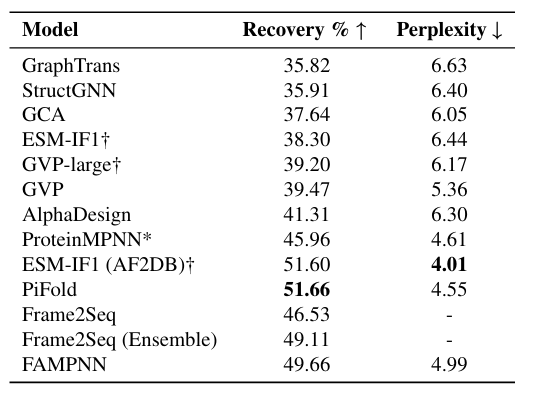
Experiments show thatFAMPNN outperforms the current state-of-the-art methods in terms of single-step sequence recovery accuracy, reaching 49.66%.In comparison, ProteinMPNN is only 45.96% and GVP is only 39.47%.
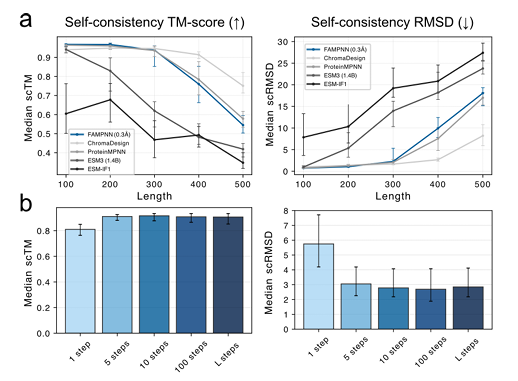
In the self-consistency evaluation on the new main chain generated based on RFdiffusion,FAMPNN (0.3Å) is comparable to ProteinMPNN in terms of scTM (structural similarity) and scRMSD (root mean square deviation) metrics.And 10 steps of iterative sampling can achieve a high degree of self-consistency, which is more efficient than the full autoregressive method. As shown in the following figure:
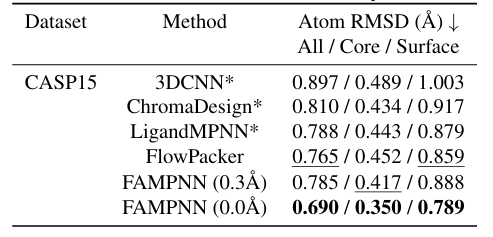
In terms of sidechain packing, the researchers compared the proposed model with other methods on the CASP13, 14, and 15 datasets.Experiments show that in the crystal structure evaluation of the CASP15 test set, the atomic RMSD (All/Core/Surface) of FAMPNN (0.0Å) is 0.690/0.350/0.789Å, which is better than other methods.And it has a strong correlation with the error of each atom and the error of each residue, with Spearman correlation coefficients of 0.843 and 0.780 respectively. As shown in the figure below:
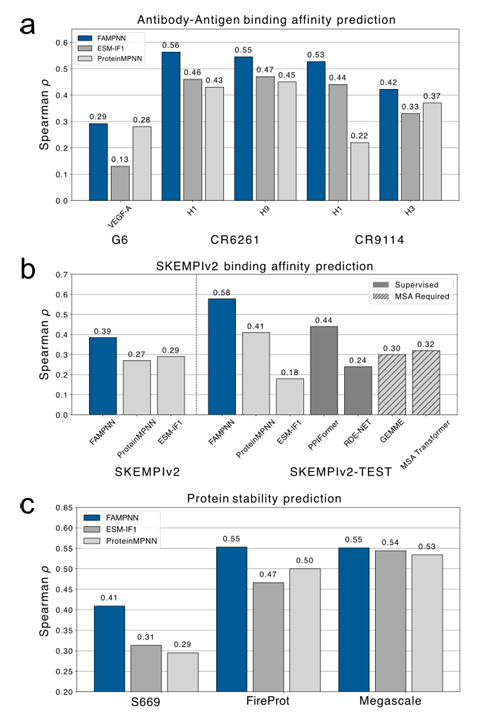
In the assessment of protein fitness under all-atom conditions,On the SKEMPlv2 dataset, FAMPNN significantly outperformed the unsupervised model, and even surpassed the supervised model on the test subset, showing strong generalization ability in zero-sample prediction. On the three stability datasets of S669, Megascale, and FireProtDB, FAMPNN performed slightly better than ProteinMPNN and ESM-IF; in the prediction of antibody-antigen binding affinity, FAMPNN always outperformed the most advanced unsupervised methods ProteinMPNN and ESM-IF1, which proves the utility of FAMPNN in protein stability and enhancing protein-protein interactions. As shown in the following figure:
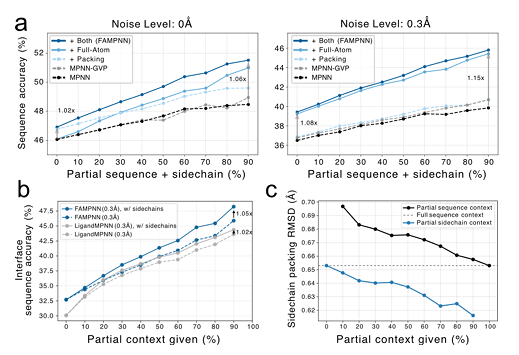
In experiments evaluating whether all-atom modeling can improve sequence design performance,The study found that adding side chain packing targets and all-atom condition settings can improve sequence accuracy.Moreover, the performance of both FAMPNN and the baseline model will improve as more structural information is injected. At the protein-protein interface, the modeling of side chain interactions is more critical, and providing partial side chain context in conjunction with the sequence can significantly improve the accuracy compared to providing only partial sequence context.
In addition, compared with LigandMPNN, FAMPNN can more effectively utilize the side chain context and perform side chain packing based on different numbers of partial sequence or side chain conformation contexts. The more contexts, the higher the packing accuracy. As shown in Figure c below:
In summary, the above experiments demonstrate that FAMPNN has significant advantages in protein fitness prediction compared with models based only on the main chain.
Driven by artificial intelligence, academia is flourishing in the field of sidechain modeling
As mentioned at the beginning, the side chain conformation is crucial to protein function. However, after the main chain of the protein is determined, there are still many possible side chain conformations, which makes the modeling and research of side chain conformations a difficult problem that must be overcome. In addition to this study, many academic research institutions around the world are driving the research of side chain modeling forward through cutting-edge deep learning technology and biological knowledge.
A team from Fudan University in China proposed a two-stage side chain modeling method called OPUS-Rota5.This method uses an improved 3D-Unet to capture local environment features.Including ligand information for each residue, and then using the RotaFormer module to aggregate various types of features. Evaluations on test sets including CAMEO and CASP15 show that OPUS-Rota5 significantly outperforms some other leading side-chain modeling methods. The related research was published on ScienceDirect with the title "OPUS-Rota5: A highly accurate protein side-chain modeling method with 3D-Unet and RotaFormer".
Paper address:
https://www.sciencedirect.com/science/article/pii/S0969212624001266
A team from Peking University proposed another method, called GeoPacker.This method combines geometric deep learning with ResNet to model protein side chains. GeoPacker explicitly represents atomic interactions in a rotationally and translationally invariant manner to extract relative position information. In terms of side chain structure prediction accuracy, GeoPacker outperforms the most advanced methods based on energy functions, and runs about 10 times and 700 times faster than the deep learning-based methods DLPacker and OPUS-Rota4, respectively, with comparable prediction accuracy. The related research was published as "GeoPacker: A novel deep learning framework for protein side-chain modeling".
Paper address:
https://onlinelibrary.wiley.com/doi/epdf/10.1002/pro.4484
At the same time, the team at the University of Toronto proposed a model called FlowPacker.Its purpose is to accurately predict the specific shape of the side chain based on the known amino acid sequence and main chain structure of the protein.Compared with previous advanced methods, FlowPacker performs better in most indicators and runs faster. For example, it has advantages in angle prediction error, the closeness between the predicted angle and the true value, atomic position deviation, etc. The related research was published under the title "FlowPacker: Protein side-chain packing with torsional flow matching".
Paper address:
In general, decoding side chain conformations is crucial to the development of the life sciences field. The continuous development of artificial intelligence technology has undoubtedly promoted the rapid development of structural biology and computational biology, and has also helped domestic and foreign research institutions to achieve a flourishing academic achievement. Once these achievements are implemented from the laboratory to the application, they will inevitably cause a new round of storms in the field of life sciences and promote biological sciences and medical sciences into a new chapter.








