Command Palette
Search for a command to run...
Based on 86,000 Protein Structure Data, a Machine Learning Method Combined With Quantum Mechanics Calculations Was Used to Discover 69 New nitrogen-oxygen-sulfur Bonds

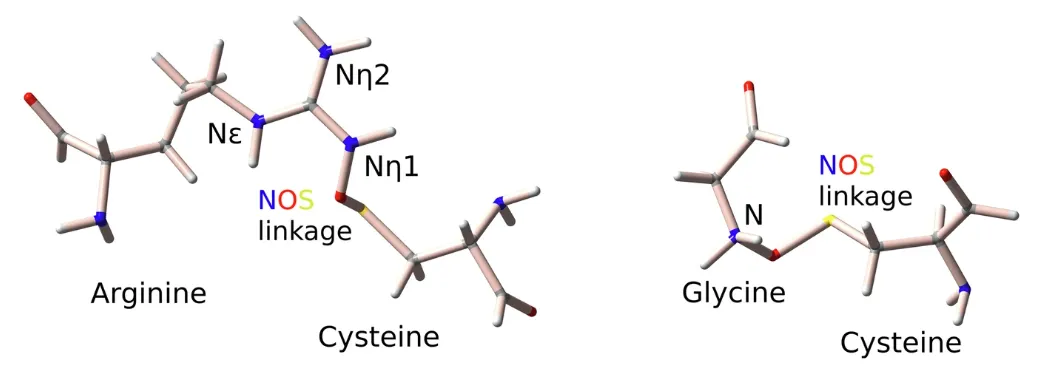
In the cell "factory", the nitrogen-oxygen-sulfur (NOS) bond is like a reversible "smart switch" that can regulate enzyme activity according to redox changes in the environment. In 2021, a team from Georg-August University of Göttingen, Germany, discovered the NOS bond between lysine and cysteine by studying the transaldolase of Neisseria gonorrhoeae.This research goes beyond the scope of single pathogen and enzyme studies and lays an important foundation for interdisciplinary protein science, drug design and bioengineering.
However, with the explosive growth of protein structure data and the continued research of the chemical bonds in protein structure by the scientific community, new problems have also arisen.Are there other NOS bonds or chemical interactions that have been overlooked?
Based on the above considerations,Sophia Bazzi and Sharareh Sayyad from George Augustus University developed an innovative computational biology algorithm, SimplifiedBondfinder.This opens a new chapter in the exploration of protein covalent bonds.The team integrated machine learning and quantum mechanics calculations to build a high-resolution X-ray crystallography database and systematically analyzed more than 86,000 high-resolution X-ray protein structures.Not only were 69 new NOS bonds discovered, but they also included novel NOS bonds formed between arginine (Arg)-cysteine and glycine (Gly)-cysteine that had never been observed before.
This revolutionary discovery broadened the scope of protein chemistry and made targeted regulation possible in drug design and protein engineering.Although this study focused on the NOS bond, the approach can be flexibly applied to study a wide range of other chemical bonds and covalent modifications.Includes structurally resolvable posttranslational modifications (PTMs).
The research results were published in Communications Chemistry under the title “Revealing arginine-cysteine and glycine-cysteine NOS linkages by a systematic re-evaluation of protein structures”.
Research highlights:
* Breaking the common scientific belief that NOS bonds only exist between lysine (Lys) and cysteine, the new redox regulation mechanism of arginine-cysteine and glycine-cysteine NOS bonds was revealed for the first time with an innovative method
* The proposed method integrates machine learning, quantum mechanical calculations and high-resolution X-ray crystallography data, solving the challenge of the lack of systematic chemical bond discovery algorithms in this field of research, breaking away from the limitations of traditional experiments, and providing a reliable and easy-to-use tool for subsequent research
* Through machine learning and artificial intelligence technologies, the cost of such research has been significantly reduced while improving research efficiency, setting an example for machine learning-driven technologies in deciphering protein functions and identifying new protein interactions

Paper address:
https://www.nature.com/articles/s42004-025-01535-w
More AI frontier papers:
https://go.hyper.ai/UuE1o
Dataset: Extracting reliable datasets with multiple layers of restrictions
The data collected by SimplifiedBondfinder comes from three different protein databases.They are PDB, PDB-REDO and BDB. The collected data will be subject to various constraints to filter out reliable and usable data sets. Among them, the database PDB-REDO (as of January 2024) refines and optimizes the static structure in the PDB to make it more in line with contemporary crystallographic standards. Compared with the original PDB entry, it has higher accuracy and reliability. As shown on the left side of the figure below:
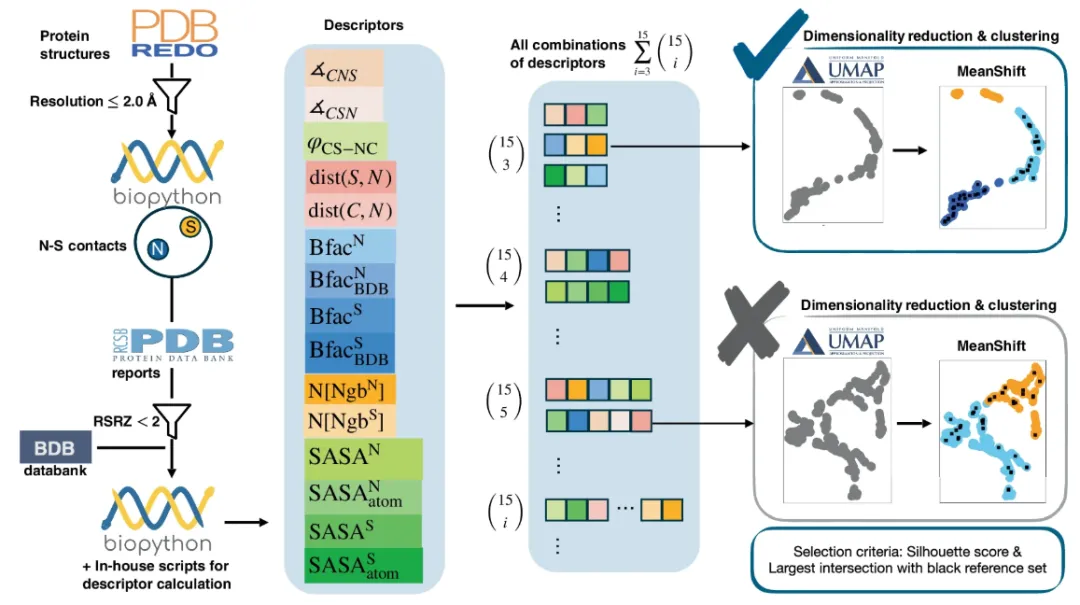
Specifically, the research team used multiple interrelated functions to drive automatic dataset generation in a database that initially contained 170,251 protein data.It first used Biopython (v 1.79) to perform structural analysis (using MMCIFParser and PDBParse) and calculate other atomic and residue properties. After only analyzing the structure determined by X-ray, the research team optimized the data of 170,127 proteins.
Subsequently, in order to further improve the prediction accuracy, the research team further screened protein structures with a resolution of ≤ 2 Å, and finally obtained 86,491 structures for experimental analysis.
To construct a data set for studying a specific chemical bond,The research team established criteria based on constituent atom types, residue names, interatomic distances, and occupancy.For NOS connections involving sulfur (S) and nitrogen (N) atoms in standard residues, the research team restricted the interatomic distance of SN, i.e., dist(S, N), to ≤ 3.2 Å, corresponding to the cutoff value for valence interactions between lysine and cysteine, and set the occupancy threshold to > 0.8 to exclude atoms with high positional uncertainty. Using this criterion, the study identified 25,462 NS contacts.
To ensure the target atomic mass depicted, the research team further applied the real-space-R-value Z-score (RSRZ) with a threshold set to <2.0 to ensure that reliable matches with the data in real space could be identified.The dataset was further reduced to 23,129 NS contacts.This allowed the experimental targets to focus mainly on two types of interactions of cysteine: the interaction between the sulfur atom of cysteine and the backbone nitrogen of glycine; and the interaction between the sulfur atom of cysteine and the side chain nitrogen of arginine and lysine.
Next,The research team used the NeighborSearch module in Biopython to extract structural parameters, collecting 15 different descriptors for each sample in each dataset.These include angles (∡CSN, ∡CNS), torsion angles (φCS-NC), other distances (dist(C, N), dist(S, N)), and the solvent accessible surface area (SASA) values of the target atoms and the corresponding residues further calculated using Bio.PDB.SASA.
The research team included atomic B-factors (Bfac) in the experiment in order to have a target atomic mobility parameter in the analysis. These values came from two databases, the RCSB PDB and a PDB file database (BDB) with consistent B-factors.
It is worth mentioning that based on the specific requirements of this study, only 15 descriptors were selected in the experiment.However, the research team said that the proposed algorithm does not have a strict limit on the number of descriptors it can process.By design it can accommodate an arbitrary number of descriptors, which enables it to integrate domain-specific knowledge or adapt to new experimental approaches.
Model architecture: integrating machine learning and quantum mechanics computing
The above part is the first step in the key steps of the proposed method, which is to construct a target data set for specific chemical bonds and apply strict criteria.This section focuses on the second key step of the proposed method, which is to use machine learning techniques to explore these high-dimensional data.Identify effective structural descriptors and predict potential sites for covalent bond formation.
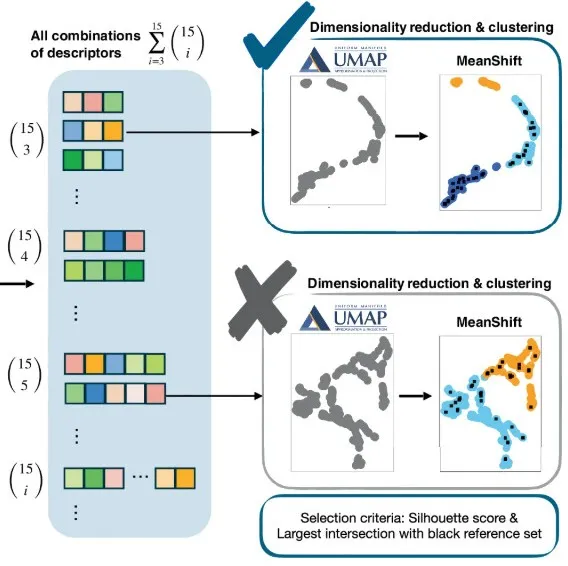
As shown in the picture above.First, the research team applied the unsupervised Uniform Manifold Approximation and Projection (UMAP) dimensionality reduction technique with a maximum embedding dimension of 3.Then mean-shift clustering is performed on all possible sets of descriptors.
in,UMAP optimally preserves the intrinsic topological and geometric properties of high-dimensional data, ensuring that essential structural features are preserved in low-dimensional embeddings.This facilitates meaningful downstream analysis. The choice of embedding dimension in UMAP depends on the topological and geometric properties of the dataset and its original high-dimensional manifold. In practical applications, two-dimensional or three-dimensional embeddings are the most interpretable because they enable intuitive visualization and assessment of clustering quality.
In this study, 3 embedding dimensions provided well-separated and meaningful clusters, justifying the choice. Chemical bond analysis and clustering results show that this dimensionality reduction method is optimal for the dataset used in this experiment. Selecting embedding dimensions higher than necessary can preserve the original popular features, but will increase computational cost without improving interpretability. Conversely, reducing the dimensionality below the optimal level will result in a large amount of information loss and poor cluster separation.
Then,The research team obtained the Silhouette Score of all three-dimensional embedding coordinates to evaluate the clustering quality of each combination.The algorithm outputs clusters, silhouette coefficients, and the reference-target connectivity within each cluster. Each candidate is identified by the name of the target atom, the corresponding residue name, residue number, chain, and PDB ID to distinguish all target atoms within the protein.
To find the final and minimal feature space, the research team used several criteria, including the value of the silhouette coefficient, the number of clusters produced by each feature space, and the distribution of reference-target connections in these clusters.
Specifically,The research team aimed to identify a feature space that can effectively segment the data into two or three distinct clusters with a silhouette coefficient ≥ 0.5.Ideally, one of the clusters does not contain any reference target connections, which is called an "impossible cluster". In practice, the minimum number of reference samples in this cluster is acceptable. The remaining clusters that contain all or most of the reference target connections are called "possible clusters".
By introducing possible and impossible candidate clusters containing the target chemical bonds,The research team was able to identify optimized feature spaces to distinguish between target atom pairs that are likely to form new chemical bonds and those that are less likely to form such bonds.Once a set of descriptors is identified that can reliably distinguish between these cases, there is no need to include additional descriptors. This approach has advantages in both computational efficiency and interpretability, and could significantly improve the predictive accuracy of methods for identifying new chemical bond formation within protein structures.
In addition to machine learning, the method proposed in this study also integrates quantum mechanical calculations.The researchers performed geometry optimization on potential candidates for NOS linkage in Lys-NOS-Cys, Gly-NOS-Cys, ARG-NηOS-Cys, and ARG-NεOS-Cys complexes. Geometry optimization was performed in water using the software package Gaussian16 – A.03 (Gaussian 16, revision C.01) at the B3LYP-D3 (BJ)/def2-TZVPD level of theory. For the optimized structures, several geometric parameters were experimentally calculated, including the distance between sulfur and nitrogen atoms (dist (S, N)), as well as the angles (∡CSN, ∡CNS, ∡NOS).
In order to verify the existence of NOS covalent bonds predicted by the proposed clustering method,The research team used phenix.refine (version 1.20.1-4487-000) to re-optimize four representative protein structures;A comprehensive structural validation was performed using phenix.molprobity to assess geometric quality, clash scores, and steric interactions to ensure consistency with high-resolution crystallographic data; a full validation report was generated using phenix.table1 summarizing refinement statistics, model quality metrics, and stereochemical deviations. These validation steps confirmed the structural integrity of the NOS junction and its compatibility with the electron density map.
Experimental results: Arg-NOS-Cys and Gly-NOS-Cys bonds are reasonable covalent bonds
To demonstrate the effectiveness of the proposed method, the research team conducted a number of experiments, exploring the use of machine learning techniques for descriptor selection, the biochemical significance of multi-descriptor space, cluster analysis, and structural and thermodynamic verification.
Selecting descriptors using machine learning
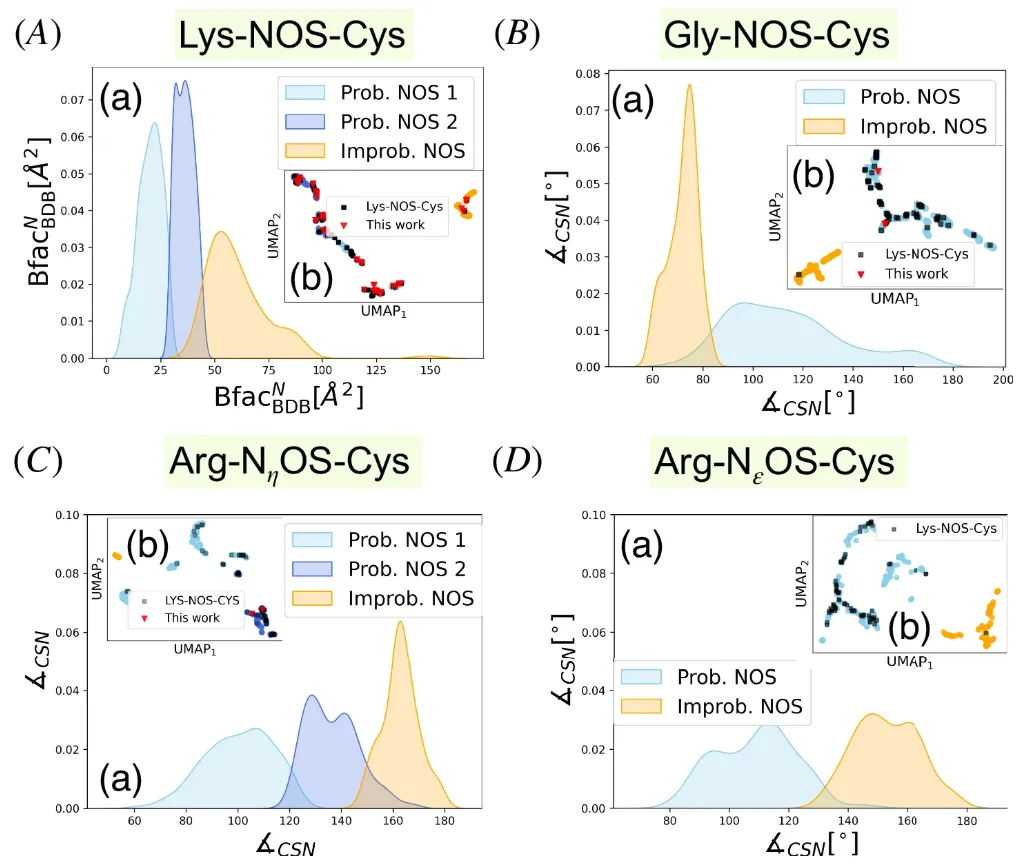
The research team first applied it to data where Lys-NOS-Cys connections were likely to exist.The dataset contains 527 lysine-cysteine pairs and also includes experimentally verified NOS bonds.The key descriptors were experimentally determined to be the B-factor of the nitrogen atom (Bfac(BDB)(N)) and the number of neighboring residues within a 4 Å radius of the Cα atoms of lysine (Ngbᴺ) and cysteine (Ngbˢ).
The research team further expanded the analysis to a dataset of 313 glycine-cysteine pairs to explore potential Gly-NOS-Cys connections, as shown in the figure below.
Here, the key descriptor sets include the B-factor of sulfur-containing residues (BfacBDBS), sulfur-nitrogen distance (dist(S,N)), and carbon-sulfur-nitrogen angle (∡CSN).
In terms of key descriptors for predicting NOS bond formation between arginine and cysteine residues,Arginine side chains have two types of nitrogen atoms, Nη and Nε, which differ in geometric characteristics and chemical properties.Therefore, we analyzed the Nη (Arg-NηOS-Cys) and Nε (Arg-Nε-Cys) datasets separately.
For Arg-NηOS-Cys, the selected descriptors correspond to the solvent accessible surface area of the nitrogen residue (SASAᴺ), ∡CSN, and the residues adjacent to sulfur (Ngbˢ) and nitrogen (Ngbᴺ); similarly for the dataset of 240 Arg-NεOS-Cys pairs, the key descriptors involve BfacBDBS, SASAˢ, the solvent accessible surface area of the nitrogen atom, ∡CSN, and ∡CNS.
These findings show clear cluster separation through UMAP dimensionality reduction visualization.As shown in the figure below, sky blue and royal blue represent NOS bond candidates, orange represents "impossible clustering", and the black squares are the reference data set. It can be clearly seen that the distribution of samples that may form NOS bonds highly overlaps with the distribution of reference standard points.
Biochemical significance of multidimensional descriptor space
The research team explored the biochemical relevance of key descriptors, which were important for distinguishing NOS from non-NOS bonds, by algorithmically determining a minimum set of descriptors.
Taking B-factor as an example, the B-factor in different clusters presents different distribution patterns. As shown in A(a) above, the modes of B-factor are different for "possible clustering" and "impossible clustering".The B-factor is related to the flexibility of atoms or regions, and active site residues usually have a lower B-factor, indicating that they are related to enzyme activity.However, the research team also pointed out that the low B-factor may indicate NOS bonding, but it may also reflect other nitrogen-sulfur interactions.
Regarding the descriptor characteristics of NOS bonds formed by different amino acid residues, BfacBDBᴺ is the main factor distinguishing the two clusters in Lys-NOS-Cys; for Gly-NOS-Cys connections, ∠CSN is the main descriptor distinguishing possible NOS connection clusters, with ∠CSN >80° for most possible samples and the ∠CSN value of the optimized Gly-NOS-Cys complex being approximately 94°; ∠CSN is still the key determinant for distinguishing possible from impossible NOS connections for Arg-NεOS-Cys connections.
Cluster analysis
In this evaluation, the research team detected 65 Lys-NOS-Cys bonds, 2 Gly-NOS-Cys bonds (Figures a and b below) and 2 Arg-NηOS-Cys bonds (Figures c and d below).
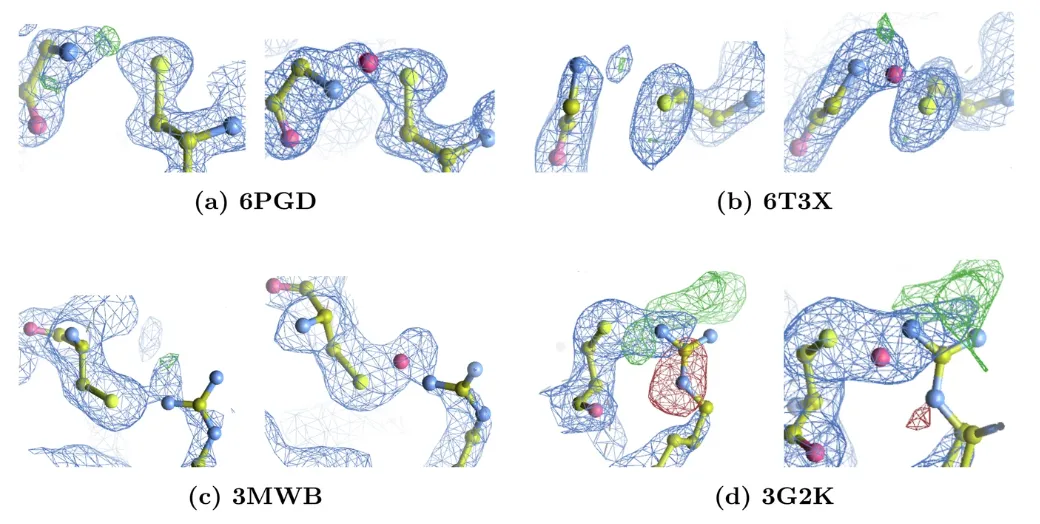
Through explicit modeling and re-refinement, the research teamAfter the introduction of NOS bonds, the Rwork/Rfree value improved by an average of 0.5%, and the unexplained electron density peak was significantly reduced.For 3G2K, there is a negative electron density peak around the arginine side chain in the original structure, which is significantly reduced after the redistribution of the arginine conformation. In addition, there are positive difference peaks near the arginine side chain in both models. Due to their large amplitude and the presence of DMSO, they may represent solvent molecules that are not modeled in the current model.
Structural and thermodynamic verification
To further confirm the connection between Arg-NOS-Cys and Gly-NOS-Cys, the research team combined quantum mechanical geometry optimization with thermodynamic evaluation of four representative protein complexes (6PGD, 6T3X, 3MWB, and 3G2K) to systematically explain the possible chemical variability in vivo.
In terms of structural verification, in the NOS bond optimized model, the SN distance ranges from 2.61 to 2.70 Å, which is very close to the 2.63 to 2.89 Å interval of the original PDB-REDO structure.Simulations with the bridging oxygen atom removed resulted in a significant increase in the SN separation to 3.36-4.26 Å, indicating that the shorter SN distances observed experimentally are consistent with the presence of an intermediate oxygen atom.
In terms of thermodynamic evaluation, the research team calculated the Gibbs free energy (ΔG) under different protonation states, showing that all NOS bond formation processes are negative.This suggests that replacing one hydrogen with an oxygen to form the NOS bond is thermodynamically feasible in the simulated state. However, the magnitude of ΔG differs significantly with protonation state and between the arginine- and glycine-derived complexes. In both systems, neutral glycine or arginine is favored over the positively charged states. The glycine-based complexes exhibit slightly higher ΔG values. While these values still imply a thermodynamically favorable association, they are systematically less exergonic than the corresponding arginine complexes.
Taken together, these structural results provide consistent evidence thatIt was shown that the Arg-NOS-Cys and Gly-NOS-Cys bonds were reasonable covalent bonds rather than simple non-bonded contacts.At the same time, the agreement between the quantum mechanically optimized geometries and the crystallographic data of the crystal system, as well as the negative free energies of formation, strongly suggest that these connections are structurally and energetically feasible in the relevant protein environment.
Machine learning opens new chapter in the microscopic world of proteins
As mentioned in the paper, the rapid development of machine learning and artificial intelligence technologies has demonstrated superiority over traditional biochemical methods in solving complex problems in biochemistry. With its low computing cost and high efficiency, it has prompted the scientific research community to launch a major revolution in "production methods" and also promoted machine learning-driven technologies to exert greater potential in deciphering protein functions and identifying new protein interactions.
Coincidentally, Kevin K. Yang et al. from California Institute of Technology published an article titled "Machine learning-guided directed evolution for protein engineering" in Nat. Methods.By comparing directed evolution and machine learning-assisted directed evolution, the superiority of machine learning is explained.At the same time, the article also lists practical cases such as enzyme catalytic efficiency and cytochrome P450 thermal stability optimization, and mentions a variety of machine learning methods such as linear regression, Gaussian process, and Bayesian optimization.It shows that machine learning can provide "data-driven intelligent navigation" for protein engineering.By modeling sequence-function relationships, the efficiency and success rate of directed evolution can be significantly improved.
Paper address:
https://arxiv.org/pdf/1811.10775
In addition, an article published by Rita Casadio et al. from the University of Bologna in Italy titled "Machine learning solutions for predicting protein-protein interactions" also detailed the exploration of machine learning in protein research.It introduces the application of machine learning methods including unsupervised and supervised learning in protein-protein molecular interactions (PPI).Key issues in data quality, representation, training algorithms, and validation procedures are highlighted.
Paper address:
https://wires.onlinelibrary.wiley.com/doi/full/10.1002/wcms.1618
In general, there are still many codes related to life hidden in the microscopic world of protein, and the systematic data-driven method with machine learning as the main means is undoubtedly like a key to open the door to the microscopic world of protein, inspiring the scientific research community to conduct more in-depth research and exploration of protein function and stability, thereby constantly breaking the limitations of human cognition of life.








